This quiz is for registered users only.



Neurotoxins may be synthetic or endogenous compounds derived from species as diverse as bacteria, fungi, spiders, marine life, and man. Seven botulinum neurotoxin (BoNT) serotypes (A, B, C, D, E, F, and G), which are produced by Clostridium botulinum Clostridium species, inhibit neurotransmitter release from nerve terminals. These serotypes of BoNT are antigenically dissimilar, utilizedistinct but related mechanisms of action, and are not interchangeable. Only BoNT serotypes A and B are approved by the Food and Drug Administration for treatment of neurological disorders and for cosmetic indications.
Neuroexocytosis, a multistage process leading to the fusion of synaptic vesicles with the plasma membrane, involves proteins collectively called SNAREs (soluble N-ethylmaleimide–sensitive factor [NSF] attachment protein receptors). Following calcium entry into the nerve terminal, 3 SNAREs form a highly stable SNARE complex which is required for fusion of synaptic vesicles with the inner surface of the plasmalemma. Membrane fusion allows the subsequent release of acetylcholine (ACh) from synaptic vesicles into the neuromuscular synaptic cleft, resulting in an action potential in the muscle that causes it to contract. After BoNT binds to its receptor and is internalized into the nerve terminal, the BoNT proteolytically cleaves its SNARE substrate, thereby blocking neuroexocytosis. Although it is generally assumed that the effects of BoNTs are restricted to the peripheral nervous system, studies suggest that BoNTs, especially at high doses, may affect higher structures in the brain. BoNT may also alter the excitability of central neural circuits, both at spinal and cortical levels, by modulating peripheral sensory inputs.
The engineering of BoNTs is a crucial step in the evolution of neurotoxins, both as research tools and for clinical therapy. Modifying the pharmacological properties of neurotoxins through protein engineering may expand and improve the efficacy of future of neurotoxin-based therapies. Through the use of recombinant technology, combining advantageous therapeutic features of each serotype has led to the development of a chimeric recombinant toxin that effectively blocks the release of pain peptides. For example, targeting a chimera of BoNT-E and BoNT-A to nociceptive neurons is a potential new therapy for pain.
The effects of BoNT occur 2 to 5 days after injection and can last 3 months or longer, but they wear off gradually as a result of pharmacokinetic and intracellular events. To achieve the best possible outcome, treatment with BoNT should be tailored to the individual needs of the patient. Understanding the mechanism of action of BoNT-A has led to the worldwide treatment of more than 100 human conditions due to hyperactivity of nerves supplying various muscles or glands.
III. INTRODUCTION
Botulinum neurotoxin (BoNT), which has been long associated with food poisoning—a form of muscle paralysis known as botulism—is now used in the clinical setting for treating neurological disorders and for cosmetic indications. BoNT inhibits cholinergic transmission at neuromuscular and autonomic postganglionic synapses. Targeted administration leads to muscle relaxation or reduction in glandular secretions, with clinical effects manifesting within 2 to 5 days and persisting for many months. Seven BoNT serotypes (A, B, C, D, E, F, and G) are produced by Clostridium botulinum, an anaerobic gram-positive bacterium, and other Clostridium species. All of these BoNT serotypes are metalloproteinases that inhibit acetylcholine (ACh) release from nerve terminals; however, their intracellular substrates, the targets of their actions, and their potencies vary substantially. BoNT-A is the most widely studied serotype for clinical purposes.1,2,
Only BoNT serotypes A and B have been approved by the Food and Drug Administration (FDA) for clinical use. Four formulations of BoNT are FDA approved (see Table 1): onabotulinumtoxinA (Botox®), abobotulinumtoxinA (Dysport™), incobotulinumtoxinA (Xeomin®), and rimabotulinumtoxinB (Myobloc®). In addition to the indications listed in Table 1, BoNT has been found to be useful in the clinical management of urological, musculoskeletal, dermatological, and secretory disorders, as well as pain and chronic migraine. Research into these and other areas is ongoing.
Table 1. FDA-approved Botulinum Neurotoxins (BoNTs)
| BoNT | Product | FDA-approved Indication(s) |
|
onabotulinumtoxinA Initial FDA approval: 1989 ------------------------------------------------- |
Botox®
------------------- |
|
| onabotulinumtoxinA
Initial FDA approval: 2002 |
Botox® Cosmetic |
|
| abobotulinumtoxinA
Initial FDA approval: 2009 |
Dysport™ |
|
| incobotulinumtoxinA
Initial FDA approval: 2010 |
Xeomin® |
|
| rimabotulinumtoxinB
Initial FDA approval: 2000 |
Myobloc® |
|
* First FDA-approved indication
Note: Only BoNT serotypes A and B have been approved by the FDA for clinical use.
Sources:
- Botox prescribing information, Allergan, Inc.
- Botox Cosmetic prescribing information, Allergan, Inc.
- Xeomin prescribing information, Merz Pharmaceuticals, LLC.
- Myobloc prescribing information, Solstice Neurosciences, Inc.
Neurological Control of Muscle Contraction
Motoneurons
Skeletal muscles are innervated by motoneurons that have their cell bodies in the brainstem or spinal cord. Motoneuron axons extend out from the central nervous system (CNS) to form peripheral nerves that branch within skeletal muscle into terminals and contact several striated muscle fibers to form neuromuscular synapses. A group of striated muscle fibers innervated by a single motoneuron constitutes a motor unit. The signal sent to a muscle to contract originates in the CNS and travels as an action potential down the motoneuron to the skeletal muscle fibers.3
Muscle Spindle Afferents
Control of body posture and movement is supported by afferent information from the proprioceptive system and mediated primarily via afferent input from 2 types of receptors located in skeletal muscles. These are: a) the muscle spindle (Figure 1),4 which is localized within the skeletal muscle and senses muscle length, and b) Golgi tendon organs, which are localized in the tendon and measure tendon tension.5
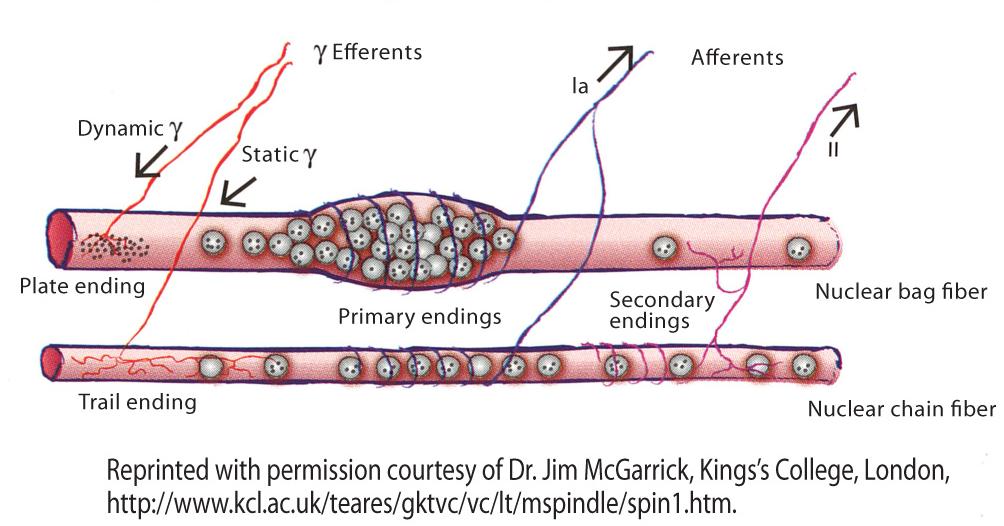
Figure 1. Muscle spindle organ.
The spindle senses muscle length and changes in length via sensory nerve terminals whose discharge rate increases as the sensory ending is stretched. Nerve terminals from type Ia afferents (shown in blue) and nerve terminals from type II afferents (shown in pink) are wrapped around specialized muscle fibers (intrafusal fibers) that are separate from the extrafusal fibers that make up the bulk of the muscle. As the muscle stretches, the spindle stretches. The activation of fusimotor fibers causes contraction of intrafusal fibers and activation of the annulospiral ring at its center. Group Ia afferents are the fastest in the body, and they register the velocity of length changes more than lengthening alone. On either side of the central area, the intrafusal fibers are able to contract if their motor supply is active. The motor supply (shown in red) comes via efferent (fusimotor) fibers that areusually classified as gamma motoneurons based on the diameter of their axons.4
Alpha (a) motoneurons innervate force-generating extrafusal muscle fibers at neuromuscular junctions, whereas gamma (?) motoneurons innervate the intrafusal muscle fibers found in muscle spindles, where they modulate the sensitivity of muscle spindles to stretch.5 In addition, most a-motoneurons receive direct group Ia–derived proprioceptive sensory input, whereas gamma motoneurons lack direct input from proprioceptive sensory afferents.6,7 When muscle stretch occurs, afferent signals from muscle spindle organs excite a-motoneurons of the stretched muscle as well as interneurons, inhibiting the motoneurons of antagonistic muscles. Signals from muscle spindle afferents are also relayed to supraspinal structures involved in long latency responses to the stretch reflex and in the generation of a body image in space. Information from tendon organs is used in part to limit forces on the tendons.5-7
IV. MECHANISMS OF ACTION OF BOTULINUM NEUROTOXINS
Botulinum Neurotoxins: Serotypes and Structures
Serotypes of BoNT are antigenically dissimilar, involve different mechanisms of action, and are not interchangeable. The 7 BoNTs are produced by Clostridium botulinum as single chain proteins and activated when proteolytically cleaved to yield a heavy chain (100-kD) and a light chain (50-kD) connected by a single disulfide bond and noncovalent bonds.8,9 Together, the heavy and light chains contain 3 functional moieties. The former is composed of the binding and translocation domains. In 1998, Stevens’ group determined the crystal structure of the entire 1,285 amino acid neurotoxin (Figure 2A).10 Approximately 10 years later, the crystal structure of the BoNT-A binding domain in complex with its lipid receptor was published (Figure 2B).11
Figure 2A. Crystal structure of BoNT-A.
The x-ray structure of the full-length BoNT-A neurotoxin was determined at 3.3 A resolution by Stevens’ group at the University of California. From Lacy et al.10 Permission pending.
Figure 2B.
Crystal structure of the BoNT-A binding domain in complex with the polysaccharide moiety of the ganglioside GT1b. Ribbon representation of the BoNT-A binding domain in rainbow color representation, from the N-terminus (blue) to the C-terminus (red). The GT1b polysaccharide is shown as yellow sticks. From Stenmark et al.11 Permission pending.
Effects of Botulinum Neurotoxins on Muscle Neuromuscular Transmission
An action potential depolarizes the motoneuron terminal to stimulate release, via exocytosis, of ACh from the lumen of synaptic vesicles into the neuromuscular synaptic cleft. When ACh reaches the postsynaptic muscle membrane, its binding to nicotinic cholinergic receptors opens a transmembrane channel, resulting in an influx of sodium ions (Na+) into the muscle fiber, followed by an efflux of potassium (K+); this initial reduction in the membrane potential of the muscle fiber generates an endplate potential which, at a threshold, creates an action potential in the muscle that causes it to contract. Nicotinic receptors are found in a variety of tissues, including the autonomic nervous system, neuromuscular junctions, and the brain of vertebrates. Nicotinic responses are excitatory, of fast onset and short duration.
Neuroexocytosis is a multistage process that leads to the fusion of synaptic vesicles with the plasma membrane. It involves proteins collectively called SNAREs (soluble N-ethylmaleimide–sensitive factor [NSF] attachment protein receptors).8 The 3 main SNAREs implicated in neuroexocytosis are synaptobrevin I/II, also known as vesicle-associated membrane protein (VAMP), syntaxin 1A/B, and synaptosomal protein 25 (SNAP-25).8 Following calcium (Ca2+) entry at the nerve terminal via depolarization-activated Ca2+ channels, these 3 SNARE proteins form a highly stable SNARE complex which is required for membrane fusion of synaptic vesicles with the inner surface of the plasma membrane. Membrane fusion allows the subsequent release of ACh from the synaptic vesicles (Figure 3A).8,12
Figure 3. BoNT and neurotransmitter release at the neuromuscular junction. p (A) Neuroexocytosis of ACh involves proteins collectively called SNAREs. The 3 main SNAREs implicated in neuroexocytosis are synaptobrevin II (also known as vesicle-associated membrane protein), syntaxin 1A/B, and SNAP-25. Following Ca2+ entry at the nerve terminal via depolarization-activated Ca2+ channels, these 3 SNARE proteins form a highly stable SNARE complex, which is required for membrane fusion of synaptic vesicles with the plasma membrane. This fusion allows the subsequent release of ACh from the synaptic vesicles.
(B) BoNT enters the neuron by binding cell surface receptors. When BoNT is injected into a target tissue, the heavy chain of the BoNT binds selectively to membrane receptors on the outer surface of the cell and is internalized into the nerve terminal via synaptic vesicle endocytosis. The Zn2+-dependent endoprotease activity of the BoNT-A light chain cleaves SNAP-25 and prevents the formation of an active SNARE complex, thereby, blocking ACh exocytosis and nerve-induced muscle contraction. From Arnon et al.12Adapted with permission from JAMA, February 2008, 2001 - Vol 285, No. 8; p 1061.
Copyright © 2001, American Medical Association. All rights reserved.
The mechanism by which BoNTs bind to nerve cell membranes involves a double-receptor model, in which the co-receptor comprises a ganglioside and a protein component.13 The binding of BoNTs to the peripheral neuromuscular junction involves the tight association between BoNTs with complex polysialogangliosides that are known to be enriched in neurons.9 Disialogangliosides (GD1b) and trisialogangliosides (GT1b) exhibit BoNT-binding affinities in the nM range and establish an initial anchorage to the neuronal membrane. BoNT serotypes A, B, C, and F bind GT1b (Figure 2B), GD1b, and GD1a. BoNT-E binds GT1b and GD1a. BoNT-G recognizes all gangliosides, and BoNT-D binds phosphatidylethanolamine.9 BoNT enters neurons by binding cell surface receptors via its heavy chain (Figure 3).12 Upon injection of BoNT into a target tissue, the heavy chain of the BoNT binds selectively to the lumenal domains of vesicle proteins that become exposed on the surface of the presynaptic cholinergic nerve terminals. BoNT-A binds to synaptic vesicle protein 2 (SV2)14; BoNT-B binds to synaptotagmin.15 The BoNT serotypes that exhibit highest sequence similarity share the same protein receptor: BoNT serotypes A, E, and F bind SV2; BoNT-B and BoNT-G bind Syt I and II.9 Stenmark et al. recently described the cell surface binding of BoNT-G to its receptor.16 The protein receptor(s) for BoNT-C and BoNT-D are still unknown.9
After BoNT binds to its receptor, the entire neurotoxin is internalized into the nerve terminal via synaptic vesicle endocytosis. A report by Zhang et al provided some evidence that the heavy chain of BoNT-A, upon internalization into neurons, remains localized in endosomes.17M In this study, the heavy chain and an attached dextran were labeled with 2 different fluorescent tags; microscopic evidence was provided for cytosolic translocation of the cargo but not the heavy chain. Once inside the cell, the light chain dissociates from the heavy chain and binds with high specificity and proteolytically cleaves its SNARE substrate, either preventing interaction with its SNARE partners or making the resulting SNARE complex unable to sustain membrane fusion (Figure 3B and Figure 4).12,18 Consequently, neuroexocytosis of ACh from the synaptic vesicles is blocked.19 Cleavage of the SNAREs abrogates vesicle fusion and synaptic transmission, thereby leading to the severe paralysis of botulism.9 In addition to cell surface receptor and intracellular substrates, the intrinsic Ca2+ dynamics in neurons may play a role in regulating BoNT activity.20
Table 2. Intracellular Targets of BoNT Serotypesa
| BoNT | Proteins Cleaved |
| BoNT-A | SNAP-25 |
| BoNT-B | VAMP (synaptobrevin) |
| BoNT-C | SNAP-25, Syntaxin 1 |
| BoNT-D | VAMP |
| BoNT-E | SNAP-25 |
| BoNT-F | VAMP |
| BoNT-G | VAMP |
aThe 3 synaptic proteins that form the SNARE complex are targets of BoNT action:
VAMP/synaptobrevin, SNAP-25, and syntaxin. BoNT serotypes B, D, F, and G cleave specifically at distinct, but different, peptide bonds of VAMP/synaptobrevin. On the other hand, BoNT types A, C, and E cleave SNAP-25 at different bonds in its carboxyl-terminus region, and BoNT type C in addition cleaves syntaxin.SNAP-25 = synaptosomal-associated protein 25; VAMP = vesicle-associated membrane protein.
Figure 4.
The ternary parallel coiled-coil structure formed by synaptobrevin, SNAP-25, and syntaxin determined by x-ray crystallography. The relative positions of bonds cleaved by BoNTs are indicated, though formation of the complex protects SNAREs against proteolysis. Synaptobrevin SNARE motif is in blue and syntaxin is green. The two SNARE motifs of SNAP-25 are represented in red and green, and are connected by an unstructured segment not present in the original crystallographic structure. From Sutton et al.18 Permission pending.
Target proteins vary among the BoNT serotypes (Table 2 and Figure 4). The Zn2+-dependent endoprotease activity of the light chain of BoNT-A causes irreversible blockade of ACh exocytosis by specific cleavage of SNAP-25, while BoNT serotypes B, D, F, and G cleave VAMP. 8,9 BoNT-C is unique among the BoNTs because it cleaves both SNAP-25 and syntaxin.8
The assumed 3-dimensional structure of BoNTs also varies among serotypes, which may explain their different behavior in terms of long-range effects.21 The effects of BoNT occur 2 to 5 days after injection and can last 3 months or longer, but they wear off gradually as a result of pharmacokinetic and intracellular catalytic events. Axonal sprouting and endplate elongation occur, but these are transient phenomena.22 The prolonged duration of effectiveness of BoNT-A compared to other serotypes may be attributable to the long lifetime of its protease activity.22
BoNT produces different effects on the muscle spindle organs. Injection of BoNT into a muscle reduces a-motoneuron activity on the extrafusal muscle fibers.23 Muscle spindles are simultaneously inhibited by the toxin’s blockade of the ?-motoneuron control of intrafusal fibers and by its subsequent reduction of Ia afferent signaling, thereby reducing feedback to the a motoneurons and other pathways to reduce muscle contraction.23,24 Injection of onabotulinumtoxinA induced atrophy in both extrafusal and intrafusal muscle fibers in the biceps femoris of Wistar rats.25 Muscle action potentials elicited by stimulation were abolished in both extrafusal and intrafusal fibers, and spindle afferent discharges were progressively reduced. Injections of onabotulinumtoxinA demonstrated that ? motoneuron terminals of isolated rat masseter muscles could be blocked, thereby reducing the Ia and II afferent signal from the muscle spindle organs and the muscle tone by reflex inhibition without affecting muscle strength.26 The effect of BoNT may, therefore, be caused by both target muscle paresis and spinal reflex inhibition.
Effect of Botulinum Neurotoxin on the Central Nervous System
Although peripherally injected BoNT appears to have no direct CNS activity, its effects on the neuromuscular junction and muscle spindle organs may affect the CNS indirectly. Current evidence suggests BoNT can alter the excitability of central neural circuits, both at spinal and cortical levels, by modulating peripheral sensory inputs. Based on studies in humans, it appears that onabotulinumtoxinA alters sensory inputs to the CNS through reversible chemodenervation of extrafusal and intrafusal fibers.27 The vibration-induced facilitation of motor-evoked potentials recorded in the sternocleidomastoid muscle in 20 healthy subjects and in 10 patients with idiopathic rotational torticollis treated with abobotulinumtoxinA or onabotulinumtoxinA suggests that BoNT denervates both extrafusal and intrafusal fibers. Furthermore, this result points to a reduced primary muscle spindle input after BoNT injection that is connected to the clinical efficacy of the treatment of spasmodic torticollis.
In preclinical studies, BoNT therapy also leads to altered afferent input to the CNS as a result of the effect on muscle spindles. The release of substance P, a neuropeptide involved in vasodilation, neurogenic inflammation, and the genesis of pain, also requires the formation of a SNARE complex that is inhibited by BoNT-A.23 The neurotoxin-induced suppression of substance P observed in the neurons of embryonic rat dorsal root ganglia was most potently associated with onabotulinumtoxinA, with an inhibitory concentration that is substantially lower than the other serotypes (inhibitory concentrations [IC50] by serotype: type A, 0.05 nM; type B, ~60 nM; type C, 0.3 nM; type F, 30 nM).28 Association of this inhibition with a decrease of SNAP-25 suggests a direct effect.
BoNT has been observed to be axonally transported in a retrograde manner by central neurons and motoneurons, transcytosed to afferent synapses, and has been detected in the hippocampus of mice29; this observation remains to be confirmed. However, retrograde transport of BoNT has not been seen in human patients, as noted in recent clinical studies. OnabotulinumtoxinA injection into the upper facial muscles in adult patients with cranial dystonia resulted in relief of muscle spasms but showed no significant effects on the silent periods in the cerebral cortex that normally occur in conjunction with transcranial magnetic stimulation.30 In addition, injection of onabotulinumtoxinA into the lower and upper extremities of children with cerebral palsy failed to alter their cortical evoked somatosensory potentials while it decreased muscle spasticity.31 The therapeutic effect of botulinum toxin on muscle dystonia has been found to normalize the asymmetrical white matter in the brain hemispheres that is common in patients with cervical dystonia, suggesting that movement disorders may lead to abnormalities in white matter; correction of the disorder may resolve the cerebral aberration.32
When BoNT is injected into a target tissue, it is almost completely bound to the axon terminal. However, when BoNT-B is applied to treat cervical dystonia, small fractions of the applied BoNT are distributed systemically and substantial systemic anticholinergic side effects can be clinically detected.33 Autonomic side effects occur more often after injections with rimabotulinumtoxinB than with onabotulinumtoxinA. Mouth dryness, accommodation difficulties, conjunctival irritation, reduced sweating, swallowing difficulties, heartburn, constipation, bladder voiding difficulties, head instability, dryness of nasal mucosa, and thrush are among the common side effects. Despite some systemic distribution, direct effects of BoNT on the CNS, when injected into patients, have not been reported, particularly due to its size (150 kDa cannot penetrate the blood-brain barrier in humans).
Although it is generally assumed that the effects of BoNTs are restricted to the peripheral nervous system, there is limited evidence that BoNTs, especially at high doses, can affect higher structures in the brain.34 Studies in cats indicate a dose-dependent effect of BoNT-A on brainstem circuitry.35,36, Moreover, there are data supporting a direct central action of BoNT-A via long-range axonal transport in motoneurons. Antonucci et al.29 demonstrated that a high dose of catalytically active BoNT-A, but not BoNT-E, undergoes retrograde axonal transport and transcytosis in different neurons. Although most of the BoNT-A effects remained restricted to the injection site, there were signs of BoNT-A activity in distant synapses. For example, cleavage of SNAP-25 was detected in the rat facial nucleus following delivery of BoNT-A to the whisker pad.29
There is clinical evidence that BoNT-A can transiently affect the excitability of cortical areas.34 Corticomotor representation was shown to be altered in patients with dystonia, and BoNT-A treatment in the dystonic limb re-established normal cortical maps.37 In subjects with upper limb dystonia, intracortical inhibition was also found to be defective, and the clinical benefit of BoNT-A correlated with a return of cortical inhibition to the levels seen in normal subjects.38, These cortical changes were completely reversible and disappeared at the completion of BoNT-A effects.
V. LONG-TERM USE OF BOTULINUM NEUROTOXIN AND ANTIBODY PRODUCTION
To achieve the best possible outcome, treatment with BoNT should be tailored to the individual needs of the patient. However, questions remain about the long-term use of BoNT and the potential for development of resistance, with repeated treatments occasionally leading to a progressive decline in therapeutic response. It has been shown that this decline may be caused by the development of neutralizing antibodies against BoNT that block its biological activity.39 These neutralizing antibodies are serotype specific, as opposed to antibodies cross-reactive against a number of antigens. Formation of antibodies after repeated treatments can reduce the duration of action and extent of maximal therapeutic effect of subsequent BoNT applications.40 Duration of action varies between patients suffering from the same condition and between those suffering from different conditions. In the same patient with identical treatment parameters, the duration of action is typically consistent.
The functionally relevant antigenicity of a BoNT preparation depends upon the amount of BoNT presented to the immune system, which is, in turn, determined by the specific biological activity and the relationship between the biological activity and the amount of total BoNT contained in the preparation.41 It is important to emphasize that essentially all the studies published on the presence of antibodies have been based on the use of the original onabotulinumtoxinA formulation. The current formulation has a much lower protein load and is, therefore, associated with a greatly reduced incidence of antibody production.42,43 Long-term immunogenicity of BoNT-B in patients with cervical dystonia has also been described.44,45 Results of recent long-term studies suggest that even though resistance to onabotulinumtoxinA is a clinical possibility, it is not a significant concern in light of the appropriate and safe use of the currently available formulations.46 It is important to note that a lack of response to a particular injection does not necessarily indicate that the patient has developed blocking antibodies. In fact, a patient may respond at a subsequent visit to exactly the same dose injected into the same muscles. Because of a certain amount of overlap between epitope regions of neutralizing antibodies to onabotulinumtoxinA or abobotulinumtoxinA and rimabotulinumtoxinB, it is not advisable to switch serotypes in clinical practice or perform simultaneous injection with both serotypes. Use of the smallest effective dose of BoNT at intervals of no less than 3 months appears to reduce the incidence of antibody formation.
Dr. Naumann discusses antigenicity.
VI. BOTULINUM NEUROTOXINS: DRUG DISCOVERY AND DEVELOPMENT
The Design of New Neurotoxins: Botulinum Neurotoxin Chimeras (Hybrid Neurotoxins)
The engineering of BoNTs is a crucial step in the evolution of neurotoxins as both a research tool and clinical therapy.20,47,48 Modifying the pharmacological properties of neurotoxins through protein engineering is believed to expand and improve the efficacy of future neurotoxin-based therapies.49,50
A pivotal study by Wang et al provided proof of principle for tailoring the pharmacological properties of BoNTs by protein engineering.47 Exchanging the C-terminal heavy chain portions of BoNT-A and BoNT-E produced 2 single-chain chimeras: chimera EA and chimera AE. This study indentified distinct domains from BoNT-A and BoNT-E that are responsible for binding and entry into neurons, cleavage of SNAP-25, and blockade of neurotransmission.47 Moreover, the mechanism underlying the rapid action of BoNT-E was revealed by using BoNT-A and –E chimeras.
Dr. Dolly discusses the development of chimeric recombinant neurotoxins
An important example of how BoNT engineering technology can lead to new and more effective neurotoxin-based therapies is the distinct effects of BoNT-A and BoNT-E on sensory neurons and the implications for the treatment of chronic pain syndromes. BoNT-A and BoNT-E, when used alone, are ineffective in blocking the release of pain mediators elicited by capsaicin from sensory neurons.8,20 However, through the use of recombinant technology, combining advantageous therapeutic features of each serotype has led to the development of a chimeric recombinant toxin that effectively blocks the release of pain peptides, such as the proinflammatory calcitonin gene-related peptide (CGRP) (Figure 5).8,20 Capsaicin activates the transient receptor potential vanilloid receptor type 1 (TRPV1) and evokes the release of CGRP. This process is dependent on specific amino acid residues of SNAP-25.20 BoNT-A binds to its receptor and is internalized and translocated to the cytosol, where it cleaves 9 residues from SNAP-25.8 However, BoNT-A does not effectively block capsaicin-induced release of CGRP from sensory neurons,8,20 because the 9 residues that BoNT-A cleaves from SNAP-25 are not required for CGRP release triggered by the unique second messenger signaling elicited by capsaicin.20 In contrast, BoNT-E protease cleaves off 26 residues from SNAP-25 that are required for capsaicin-induced CGRP release. However, BoNT-E is not effectively internalized by sensory neurons due to the limited number of BoNT-E receptors on the surface of these cells and, therefore, cannot block capsaicin-induced CGRP release. A study by Meng et al20,51 showed that a recombinant chimera of BoNT-A and BoNT-E (-EA) attenuated TRPV1-mediated CGRP release. CA2+ -EA chimera contained the BoNT-A binding domain and the BoNT-E protease domain, and was effectively internalized by sensory neurons and inhibited CGRP release in vitro and in situ. Thus, targeting -EA chimera to nociceptive neurons is a potential new therapy for pain.
Dr. Dolly discusses the antinociceptive properties of neurotoxins.
Figure 5.
A chimera BoNT-E and BoNT-A blocks the release of pain peptides.Illustration shows a trigeminal ganglionic neuron (TGN). TGNs predominantly express SV2C, the receptor for BoNT-A, but not SV2A/B, which act as main receptors for BoNT-E. BoNT-A binds the TGN and enters synaptic vesicles, where the BoNT-A light chain protease is delivered into the cytosol and cleaves SNAP-25. Truncated SNAP-25 causes a moderate destabilization of the SNARE complex (SNAP-25, syntaxin, and VAMP), which leads to the inhibition of CGRP release evoked by K+ depolarization and Ca2+ entry. However, BoNT-A–induced cleavage of SNAP-25 does not inhibit capsaicin-induced CGRP release. The -EA chimera binds to the SV2C receptor via the heavy chain domain of BoNT-A. After the -EA chimera enters the TGNs, the BoNT-E light chain protease cleaves off a larger fragment of SNAP-25, and blocks CGRP release elicited by all stimuli. VACC=voltage-activated Ca2+ channels. From Dolly et al.8
Recombinant Neurotoxins and Therapy of Non-Neurological Disorders
The engineering of BoNT derivatives holds promise for the development of neurotoxin-based therapies for non-neurological disorders. Such therapies rely on retargeting the BoNT light chain to non-neuronal SNARE isoforms. For example, BoNT light chains may be engineered to target to non-neuronal SNAREs that regulate secretion of airway mucus, insulin, gastric acid, and ions.50 A recent study by Chen and Barbieri showed that a mutated BoNT-E protease cleaved the non-neuronal SNARE protein SNAP-23. This BoNT-E protease derivative inhibited mucin secretion in cultured human epithelial cells, suggesting that engineered BoNTs may lead to therapies for human hypersecretion diseases.50
Preventing Neurotoxin Action In Vivo
BoNTs are the most toxic substances known.52 Approximately 1,000 cases of human BoNT poisoning are reported each year worldwide.53 Moreover, BoNT has been a concern to the US military and its allies as a biowarfare weapon since World War II. More recently, the Centers for Disease Control and Prevention (CDC) recognized BoNTs as a potential bioterrorist threat to the public.54,55 Thus, strategies to prevent and treat botulism are actively sought worldwide.52 A promising DNA-based therapy for botulism is the development of recombinant fragment vaccines.54,55 Recombinant technology allows highly purified and efficacious antigens to be produced in adequate quantities without the need to culture and manipulate large volumes of BoNT. Phase II clinical trials with recombinant vaccines are under way.54,55
Small molecule compounds (<500 Da) can potentially be used to treat pre- and post-exposure BoNT intoxication by regulating the catalytic activity of BoNT enzymes. Several recent studies have identified small molecule inhibitors representing candidate structural classes for the development of drugs that could be used to inactivate the zinc protease domain of BoNT.52,53
VII. BEYOND BOTULINUM NEUROTOXIN: OTHER NEUROTOXINS
One of the first highly selective neurotoxins discovered was anti–nerve growth factor (NGF), which severely suppresses the growth and development of sympathetic noradrenergic neurons.56 Novel humanized antibodies directed against NGF are now in advanced clinical trials for the therapy of chronic pain.57 Studies in the mid-1960s characterized 6-hydroxydopamine (6-OHDA), a neurotoxin that destroys both norepinephrine- and dopamine-containing neurons in the brain. Later, neurotoxins that were selective for glutamate receptor subtypes were either discovered or developed.58
Neurotoxins may be synthetic (eg, cocaine) or endogenous compounds; the latter come from species as diverse as bacteria, fungi, spiders, marine life, and man. For example, the tryptophan metabolite quinolinic acid is an endogenous neurotoxin produced in the human brain, and its levels may be altered adversely by infection or environmental factors.58 Indeed, the repertoire of bioneurotoxins extends well beyond BoNTs. Tetanus neurotoxin, like BoNT, is produced by bacteria of the genus Clostridium and consists of 2 disulfide-linked heavy and light chains. Whereas botulism is caused by the inhibition of ACh release at peripheral synapses, tetanus is due to the persistent inhibition of neurotransmitter release at central inhibitory synapses.59,60 Puffer fish poisoning is due to a neurotoxin called tetrodotoxin, which is produced by marine bacteria that live in that kind of fish.61,62
Neurotoxins from scorpions, sea anemones, and cone snails have served as research tools for the identification and functional characterization of voltage-dependent ion channels.63 a-Latrotoxin is the major neurotoxin isolated from black widow spiders. Upon binding to one of its specific presynaptic receptors, a-latrotoxin has been shown to form Ca2+-permeable pores in presynaptic membranes, allowing extracellular Ca2+ to enter the presynaptic terminal and fulminant transmitter release at autonomic synapses.63 Conotoxins are secreted by venomous marine snails of the genus Conus.64 These toxins selectively inhibit voltage-sensitive Ca2+ channels. O-Conotoxins, which are selective for neuronal (N)-type Ca2+ channels, have emerged as potential new drugs for the treatment of chronic pain.65
Dr. Dolly describes ongoing research with neurotoxins from non-botulinum sources.
REFERENCES
- 1. Jankovic J, et al. Botulinum Toxin: Therapeutic Clinical Practice and Science. Philadelphia, PA: Saunders; 2009.
- 2. Ward AB, Barnes MP. Clinical Uses of Botulinum Toxins. Cambridge University Press; 2007
- 3. Firestein GS, Budd RC, Harris ED Jr, et al, eds: Kelley's Textbook of Rheumatology. 8th ed. Philadelphia, PA: Elsevier; 2008.
- 4. King’s College Web site. http:www.kcl.ac.uk/teares/gktvc/vc/lt/msprindle/spin.1.htm.
- 5. Burke RE, Strick PL, Kanda K, Kim C, Walmsley B. Anatomy of medial gatrocnemius and soleus motor nuclei in cat spinal cord. J Neurophysiol. 1977;40:667-680.
- 6. Eccles JC, Eccles RA, Lundberg A. The convergence of monosynaptic excitatory afferents on to many different species of alpha motoneurones. J Physiol.
1957;137:22-50. - 7. Eccles JC, Eccles RM, Iggo A, Lundberg A. Electrophysiological studies in gamma motoneurones. Acta Physiol Scand. 1960;50:32-40.
- 8. Dolly JO, Lawrence GW, Meng J, Wang J, Ovsepian SV. Neuro-exocytosis: botulinum toxins as inhibitory probes and versatile therapeutics. Curr Opin Pharmacol. 2009;9:326-335.
- 9.Montal M. Botulinum neurotoxin: a marvel of protein design. Annu Rev Biochem. 2010;79:10.1-10.27.
- 10. Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat Struct Biol. 1998;5(10):898-902.
- 11.a Stenmark P, Dupuy J, Imamura A, Kiso M, Stevens RC. Crystal structure of botulinum neurotoxin type A in complex with the cell surface co-receptor GT1b-insight into the toxin-neuron interaction. PLoS Pathog. 2008;4(8):e1000129.
- 12. Arnon SS, Schechter R, Inglesby TV, et al; Working Group on Civilian Biodefense. Botulinum toxin as a biological weapon. Medical and public health management. [Consensus statement] JAMA. 2001;285(8):1059-1070.
- 13. Aoki KR, Smith LA, Atassi MZ. Mode of action of botulinum neurotoxins: current vaccination strategies and molecular immune recognition. Crit Rev Immunol. 2010;30(2):167-187.
- 14. Dong M, Yeh F, Tepp WH, Dean C, et al. SV2 is the protein receptor for botulinum neurotoxin A. Science. 2006.Apr 312(5773):540-541.
- 15. Dong M, Richards DA, Goodnough MC, et al. Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J Cell Biol. 2003;162:1293-1303.
- 16. Stenmark P, Dong M, Dupuy J, Chapman ER, Stevens RC. Crystal structure of the botulinum neurotoxin type G binding domain: insight into cell surface binding. J Mol Biol. 2010;397:1287-1297.
- 17. Zhang P, Ray R, Singh BR, Li D, Adler M, Ray P. An efficient drug delivery vehicle for botulism countermeasure. BMC Pharmacology. 2009;9:12.
- 18. Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347-353.
- 19. Dolly JO, Black J, Williams RS, Melling J. Acceptors for botulinum neurotoxin reside on motor nerve terminals and mediate its internalization. Nature. 1984;307:457-460.
- 20. Meng J, Ovsepian SV, Wang J, et al. Activation of TRPV1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti-nociceptive potential. J Neurosci. 2009;29:4981-4992.
- 21. Kumaran D, Eswaramoorthy S, Furey W, Navaza J, Sax M, Swaminathan S. Domain organization in Clostridium botulinum neurotoxin type E is unique: its implication in faster translocation. J Mol Biol. 2009;386:233-245.
- 22.De Paiva A, Meunier FA, Molgo J, Aoki KR, Dolly JO. Functional repair of motor endplates after botulinum neurotoxin type A poisoning. Proc Natl Acad Sci U S A. 1999;96(6):3200-3205.
- 23. Aoki KR, Guyer B. Botulinum toxin type A and other botulinum toxin serotypes: a comparative review of biochemical and pharmacological actions. Eur J Neurol. 2001;8(Suppl 5):21-29.
- 24. Aoki KR. Botulinum toxin: a successful therapeutic protein. Curr Med Chem. 2004;11:3085-3092.
- 25. Rosales RL, Arimura K, Takenaga S, Osame M. Extrafusal and intrafusal muscle effects in experimental botulinum toxin-A injection. Muscle Nerve. 1996;19:488-496.
- 26. Filippi GM, Errico P, Santarelli R, Bagolini B, Manni E. Botulinum A toxin effects on rat jaw muscle spindles. Acta Otolaryngol. 1993;113:400-404.
- 27. Urban PP, Rolke R. Effects of botulinum toxin type A on vibration induced facilitation of motor evoked potentials in spasmodic torticollis. J Neurol Neurosurg Psychiatry. 2004;75:1541-1546.
- 28. Welch MJ, Purkiss JR, Foster KA. Sensitivity of embryonic rat dorsal root ganglia neurons to Clostridium botulinum neurotoxins. Toxicon. 2000;38:245-258.
- 29. Antonucci F, Rossi C, Gianfranceschi L, Rossetto O, Caleo M. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28(14):3689-3696.
- 30. Allam N, Fonte-Boa PM, Tomaz CA, Brasil-Neto JP. Lack of effect of botulinum toxin on cortical excitability in patients with cranial dystonia. Clin Neuropharmacol. 2005;28:1-5.
- 31. Bockowski L, Okurowska-Zawada B, Sobaniec W, Kulak W, Sendrowski K. Cortical somatosensory evoked potentials and spasticity assessment after botulinum toxin type A injection in children with cerebral palsy. Adv Med Sci. 2007;52(Suppl 1):171-175.
- 32. Blood AJ, Tuch DS, Makris N, Makhlouf ML, Sudarsky LR, Sharma N. White matter abnormalities in dystonia normalize after botulinum toxin treatment. Neuroreport. 2006;17:1251-1255.
- 33. Dressler D, Benecke R. Autonomic side effects of botulinum toxin type B treatment of cervical dystonia and hyperhidrosis. Eur Neurol. 2003;49:34-38.
- 34. Caleo M, Schiavo G. General effects of tetanus and botulinum neurotoxins. Toxicon. 2009;54:593-599.
- 35. Moreno-Lopez B, de la Cruz RR, Pastor AM, Delgado-Garcia JM. Effects of botulinum neurotoxin type A on abducens motorneurons in the cat: alterations of the discharge pattern. Neuroscience. 1997;81(2):437-455.
- 36. Moreno-Lopez B, de la Cruz RR, Pastor AM, Delgado-Garcia JM. Botulinum neurotoxin alters the discharge characteristics of abducens motorneurons in the alert cat. J Neurophysiol. 1994;72(4):2041-2044.
- 37. Byrnes ML, Thickbroom GW, Wilson SA, Shipman JM, Stell R, Mastaglia FL. The corticomotor representation of upper limb muscles in writer's cramp and changes following botulinum toxin injection. Brain. 1998;121 (Pt 5):977-988.
- 38. Gilio F, Curra A, Lorenzano C, Modugno N, Manfredi M, Berardelli A. Effects of botulinum toxin A on intracortical inhibition in patients with dystonia. Ann Neurol. 2000;48:20-26.
- 39. Dolimbek BZ, Steward LE, Aoki KR, Atassi MZ. Immune recogntion of botulinum neurotoxin B: antibity-binding regions of the heavy chain of the toxin. Mol Immunol. 2008;45:910-924.
- 40. Dressler D. Clinical presentation and management of antibody-induced failure of botulinum toxin therapy. Mov Disord. 2004;19(Suppl 8):S92-S100.
- 41. Dressler D, Hallett M. Immunological aspects of Botox, Dysport and Myobloc/NeuroBloc. Eur J Neurol. 2006;13(Suppl 1):11-15.
- 42. Jankovic J, Vuong KD, Ashan J. Comparison of efficacy of immunogenicity of original versus current botulinum toxin in cervical dystonia. Neurology. 2003;60:1186-1188.
- 43. Yablon SA, Brashear A, Gordon MF, et al. Formation of neutralizing antibodies in patients receiving botulinum toxin type A for treatment of poststroke spasticity: a pooled-data analysis of three clinical trials. Clin Ther. 2007;29(4):683-690.
- 44. Jankovic J, Hunter C, Dolimbek BZ, et al. Clinico-immunologic aspects of botulinum toxin type B treatment of cervical dystonia. Neurology. 2006;67:2233-2235.
- 45. Atassi MZ, Dolimbeck BZ, Jankovic J, Steward LE, Aoki KR. Molecular recognition of botulinum neurotoxin B heavy chain by human antibodies from cervical dystonia patients that develop immunoresistance to toxin treatment. Mol Immunol. 2008;45:3878-3888.
- 46. Brin MF, Comella CL, Jankovic J, Lai F, Naumann M. Long-term treatment with botulinum toxin type A in cervical dystonia has low immunogenicity by mouse protection assay. Mov Disord. 2008;23:1353-1360.
- 47. Wang J, Meng J, Lawrence GW, et al. Novel chimeras of botulinum neurotoxins A and E unveil contributions from the binding, translocation, and protease domains to their functional characteristics. J Biol Chem. 2008;283:16993-17002.
- 48. Band PA, Blair S, Neubert TA, Cardozo TJ, Ichtchenko K. Recombinant derivatives of botulism neurotoxin A engineered for trafficking studies and neuronal delivery. Protein Expr Purif. 2010;71:62-73.
- 49.Muraro L, Tosatto S, Motterlini L, Rossetto O, Montecucco C. The N-terminal half of the receptor domain of botulinum neurotoxin A binds to microdomains of the plasma membrane. Biochem Biophys Res Commun. 2009;380:76-80.
- 50. Chen S, Barbieri JT. Engineering botulinum neurotoxin to extend therapeutic intervention. Proc Natl Acad Sci U S A. 2009;106:9180-9184.
- 51. Meng J, Wang J, Lawrence G, Dolly JO. Synaptobrevin I mediates exocytosis of CGRP from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. J Cell Sci. 2007;120:2864-2874.
- 52. Lai H, Feng M, Roxas-Duncan V, Dakshanamurthy S, Smith LA, Yang DC. Quinolinol and peptide inhibitors of zinc protease in botulinum neurotoxin A: effects of zinc ion and peptides on inhibition. Arch Biochem Biophys. 2009;491:75-84.
- 53. Roxas-Duncan V, Enyedy I, Montgomery VA, et al. Identification and biochemical characterization of small-molecule inhibitors of Clostridium botulinum neurotoxin serotype A. Antimicrob Agents Chemother. 2009;53:3478-3486.
- 54. Webb RP, Smith TJ, Wright P, Brown J, Smith LA. Production of catalytically inactive BoNT/A1 holoprotein and comparison with BoNT/A1 subunit vaccines against toxin subtypes A1, A2, and A3. Vaccine. 2009;27:4490-4497.
- 55. Smith LA. Botulism and vaccines for its prevention. Vaccine. 2009;27(Suppl 4):D33-D39.
- 56. Kessler JA. Parasympathetic, sympathetic, and sensory interactions in the iris: nerve growth factor regulates cholinergic ciliary ganglion innervation in vivo. J Neurosci. 1985;5(10):2719-2725.
- 57. Cattaneo A. Tanezumab, a recombinant humanized mAb against nerve growth factor for the treatment of acute and chronic pain. Curr Opin Mol Ther. 2010;12:94-106.
- 58.Kostrzewa RM. Evolution of neurotoxins: from research modalities to clinical realities. Curr Protoc Neurosci. 2009;Chapter 1:Unit 1.18.
- 59. Tonello F, Morante S, Rossetto O, Schiavo G, Montecucco C. Tetanus and botulism neurotoxins: a novel group of zinc-endopeptidases. Adv Exp Med Biol. 1996;389:251-260.
- 60.Lalli G, Bohnert S. Deinhardt K, Verastegui C, Schiavo G. The journey of tetanus and botulinum neurotoxins in neurons. Trends Microbiol. 2003;11(9):431-437.
- 61. Noguchi T, Arakawa O. Tetrodotoxin—distribution and accumulation in aquatic organisms, and cases of human intoxication. Mar Drugs. 2008;6:220-242.
- 62. Chamandi SC, Kallab K, Mattar H, Nader E. Human poisoning after ingestion of puffer fish caught from Mediterranean Sea. Middle East J Anesthesiol. 2009;20:285-288.
- 63. Luch A. Mechanistic insights on spider neurotoxins. EXS. 2010;100:293-315.
- 64. Wen L, Yang S, Qiao H, et al. SO-3, a new O-superfamily conopeptide derived from Conus striatus, selectively inhibits N-type calcium currents in cultured hippocampal neurons. Br J Pharmacol. 2005;145:728-739.
- 65. Berecki G, Motin L, Haythornthwaite A, et al. Analgesic (omega)-conotoxins CVIE and CVIF selectively and voltage-dependently block recombinant and native N-type calcium channels. Mol Pharmacol. 2010;77:139-148.


 |