This quiz is for registered users only.




II. ABSTRACT
Dystonia is defined as a neurological syndrome characterized by involuntary, sustained, patterned, and often repetitive muscle contractions of opposing muscles, resulting in twisting or squeezing movements, abnormal postures, or both. Dystonia can be subdivided according to anatomical distribution into focal, segmental, and generalized. Although pathogenesis-targeted therapy is not yet available, treatment strategies designed to relieve motor symptoms and pain have markedly improved the quality of life of patients with dystonia. Local chemodenervation with botulinum neurotoxin (BoNT) is considered the treatment of choice for patients with focal dystonia, whereas medications and deep brain stimulation are usually used in patients with segmental and generalized dystonia.
III. DYSTONIA OVERVIEW
Introduction
Dystonia is a heterogeneous group of movement disorders characterized by involuntary, sustained, or rapid, but patterned, muscle contractions that result in twisting or squeezing movements or abnormal postures. Classified by anatomical distribution, dystonia can be focal (affecting a single area of the body), segmental (involving 2 or more contiguous regions of the body), or generalized (involving at least 1 leg, the trunk, and another region of the body).
Patient with generalized, leg and truncal dystonia (camptocormia)
Video courtesy of Joseph Jankovic, MD with permission from patient
Common types of focal dystonia include blepharospasm; oromandibular, lingual, or facial dystonia; cervical dystonia; laryngeal dystonia (also referred to as spasmodic dysphonia); limb dystonia; and task-specific dystonias, such as writer's cramp or musician's and other occupational dystonias.1 This review focuses on cervical dystonia, spasmodic dysphonia, and focal hand dystonia; blepharospasm is discussed in another chapter.
Dystonia can be also classified by etiology as primary dystonia, usually idiopathic or genetic, and secondary dystonia, caused by an underlying neurological condition such as stroke, cerebral palsy, neurodegenerative disease, or by certain medications such as dopamine receptor blocking drugs (tardive dystonia).
Primary dystonias, by definition, are not associated with any other neurological signs, while secondary dystonias are also referred to as "dystonia plus" because of co-existent neurological deficits or disorders.
Epidemiology
The incidence and prevalence rates of dystonia are likely underestimated because only a few well-designed epidemiological studies have been conducted.2,3 According to the Epidemiological Study of Dystonia in Europe (ESDE) Collaborative Group, the prevalence of primary dystonia was estimated to be 15.2 per 100,000 persons annually, based on pooled data from 8 European countries.2 This is similar to other estimates, although some studies have reported a much higher prevalence, particularly among Ashkenazi Jews (of Eastern European ancestry).
Pathogenesis
The exact pathogenesis of dystonia remains unclear. Primary dystonia is most commonly idiopathic, although some, such as DYT1, DYT6, DYT7, and DYT13, are due to specific genetic abnormalities.4-7 By definition, primary dystonia is not associated with any neurological deficits, and routine brain magnetic resonance imaging (MRI) and other neurodiagnostic tests in these cases are normal. Primary dystonia is believed to involve reduced inhibition of thalamocortical output, resulting in simultaneous contraction of agonist and antagonist muscles.8
Dystonia is classified as secondary when it occurs in association with a lesion in the central nervous system (CNS), which can be caused by stroke, cerebral palsy, encephalitis, environmental insults, or neurodegenerative diseases. Secondary dystonia is often associated with abnormalities on neurological examination and on brain MRI.
Current hypotheses suggest that the basal ganglia, midbrain, and possibly the cerebellum play an integral role in the pathogenesis of dystonia.9 The causes for most dystonias are unknown, although as many as 15 monogenic subtypes have been identified.4,5,10 A mutation in the DYT1 or TOR1A gene, coding for torsinA, an ATP-binding protein,11 may be responsible for the most common primary genetic dystonia. DYT1 dystonia typically begins in childhood with a distal limb (hand or foot) being affected first, followed by progression to generalized dystonia. However, the phenotypic spectrum of DYT1 dystonia is broad, and onset can occur late in life.12 In a study of 170 TOR1A carriers, the mean age at onset was 17.6 years, with a range from 3 to 70 years.13 DYT1 dystonia accounts for approximately 40% to 65% of early-onset primary dystonia in non-Jewish populations and 90% of early-onset limb dystonia in the Ashkenazi Jewish population.14 Inheritance is autosomal dominant, but penetrance is only approximately 30% to 40%; thus, most mutation carriers do not express apparent disease.15
In some cases, dystonia and parkinsonism co-exist, and evidence of reduced dopamine neurotransmission in the striatum may be detected.16 PLA2G617,18 mutations, for example, have been described to cause adult-onset dystonia–parkinsonism.
Diagnosis
Appropriate diagnosis and classification of dystonia are critical for predicting the prognosis and for selecting the most appropriate treatment strategy. The diagnosis of dystonia is based mainly on clinical features, although diagnostic laboratory data can provide supportive evidence. Unfortunately, diagnosis is often delayed because the initial clinical presentation is wrongly attributed to other causes.19 Dystonias are often misdiagnosed as other conditions, and there is a lack of consensus on terminology among physicians, as well as a lack of a diagnostic algorithm.8,19 Therefore, consultation by a movement disorder specialist is suggested to confirm the diagnosis. The diagnosis of secondary dystonia should be considered when there are additional neurological abnormalities, which is why this group of disorders if often referred to as "dystonia plus."
Cervical dystonia (CD), also known as spasmodic torticollis, is the most common form of focal dystonia. CD is often a debilitating neurological movement disorder characterized by abnormal, involuntary, and often painful contractions of the cervical and/or shoulder muscles, which result in twisting and repetitive movements or abnormal postures of the neck and head.
a) Epidemiology
According to the National Spasmodic Torticollis Association (NSTA), the prevalence rate of CD in the United States is approximately 30 of every 100,000 persons.20 The prevalence of CD was estimated to be 5.7 per 100,000 according to the ESDE Collaborative Group,2 but other reviews cite figures between 3 and 732 per 100,000.21 Based on a survey of 60,062 respondents to an e-mail questionnaire, the prevalence of CD has been estimated to be 2.80 per 100,000 persons (0.28%).3 Women are more commonly affected by CD than men (2:1), and although the average age at onset is in the fourth decade, men experience a significantly earlier age at onset.22 Current hypotheses suggest that the basal ganglia, midbrain, and possibly, the cerebellum play an integral role in the pathogenesis of dystonia, including CD.9 In the majority of patients with CD, the etiology is not identifiable, and the disorder is often classified as primary focal dystonia.
b) Clinical Features
The clinical presentation of symptoms varies among patients with CD. Pain, partly related to the muscle spasm or inflammatory or degenerative changes in the cervical spine, is reported in 75% of patients and is often the cause of significant disability.23 Among early symptoms, patients usually report "pulling" or "drawing" in the neck and/or an involuntary twisting, shaking, or jerking of the head. Patients with CD typically present with head rotation or deviation (see video below), although a wide variety of abnormal head and neck postures are possible. The most common is rotational torticollis (rotation of the chin around the vertical axis toward 1 shoulder), followed by laterocollis (tilting of the head in the coronal plane, moving 1 ear toward the ipsilateral shoulder), retrocollis (backward extension of the neck), and anterocollis (forward flexion of the neck).
Cervical dystonia before and 6 months after botulinum neurotoxin treatment
Video courtesy of Joseph Jankovic, MD with permission from patient
The course of the disease varies from patient to patient. Most patients with CD report a progression in severity of symptoms in the first 5 years. Approximately one-third of patients progress to segmental dystonia. Spontaneous remissions, observed in about 20% of patients, occur more frequently during the first year and in patients with an early age of onset, but they usually are transient and in almost all cases, the dystonia recurs after a few weeks or months.
CD has a significantly negative impact on a patient's quality of life (QOL). Among those with CD, self-reported QOL was comparable to that of patients with serious neurological conditions, such as multiple sclerosis, Parkinson's disease, and stroke.24 Compared with patients who have other types of focal dystonia (eg, blepharospasm, writer's cramp), patients with CD reported a poorer QOL.25 CD appears to have a significant psychosocial impact on a patient's life. Compared with the general population, the prevalence rates of clinically relevant social phobia, mood disorders, and psychiatric comorbidity, including depressive disorders, are significantly higher in patients with CD.26,27
c) Diagnosis
A comprehensive examination of the patient's medical and family history and routine neurological examination are the important initial steps in diagnosing CD. The presence of a sensory trick is considered affirmative for a diagnosis of CD. Sensory tricks, also known as geste antagoniste, such as gently touching the face, head, or neck, are techniques typically used to relieve dystonic muscle contraction and correct the abnormal posture. The mechanism of action of geste antagoniste is unknown; however, it is hypothesized that sensory tricks involve alterations of peripheral proprioceptive feedback modifying the motor output.28
Currently, no widely accepted consensus on use of a disease-specific scale has been reached, but a variety of subjective and objective scales are available for assessing CD. The most commonly used is the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS), which consists of 3 subscales: severity, disability, and pain.29 Another frequently applied scale described by Tsui et al assesses head and shoulder positions, duration of sustained movements, and head tremor in patients with CD.30 Interrater reliability has been demonstrated with this scale.31 The Burke-Fahn-Marsden Dystonia Scale, which assesses movement and disability, is also often used.32 Recently, a patient-based rating scale, the Cervical Dystonia Impact Profile (CDIP-58), was developed and found to yield reliable and valid data in measuring the health impact of CD on head and neck symptoms, pain and discomfort, upper-limb activities, walking, sleep, annoyance, mood, and psychosocial functioning.33 Quality of life domains, such as stigma, emotional well-being, pain, activities of daily living, and social/family life, can be assessed using the Craniocervical Dystonia Questionnaire (CDQ-24) in patients with CD and blepharospasm.34
d) Management of CD
Currently available interventions for dystonia include physical therapy designed to prevent contractures; use of muscle-relaxation techniques, constrained-induced, and other rehabilitative therapies; oral medications such as levodopa, anticholinergics, and baclofen; intramuscular injections of botulinum neurotoxins (BoNTs); intrathecal infusion of baclofen; and surgical procedures, particularly deep-brain stimulation (DBS) of the globus pallidus internus (GPi).35,36 For patients with secondary dystonia, etiology-specific treatment should be sought. For example, patients in whom dystonia is caused by exposure to dopamine receptor blocking drugs (neuroleptics), so-called tardive dystonia, should have the offending drug discontinued. Similarly, patients with Wilson's disease and other metabolic disorders may require specific treatment strategies.
There is no cure for CD, but safe and effective therapies are available. The primary objective of the treatment of CD is alleviation of dystonic contractions and associated symptoms such as pain, maintenance of functional ability and QOL, and prevention of long-term complications (eg, contractures, radiculopathy, or compressive cervical myelopathy).
Oral Pharmacologic Treatment
Anticholinergic agents, dopamine receptor antagonists, and GABAmimetic agents have a low rate of efficacy in patients with CD.37 Anticholinergics are commonly used to treat focal, segmental, and generalized dystonia; however, no specific controlled trials for the treatment of CD with these agents have been conducted. Although levodopa is the treatment of choice for dopa-responsive dystonia (Segawa syndrome), no evidence supports the use of dopaminergic agents for the treatment of other primary or secondary dystonias, including CD. Even though benzodiazepines, such as clonazepam and diazepam, are used in clinical practice to treat patients with mild dystonias, no controlled trial has evaluated this therapeutic approach in patients with CD. Oral baclofen, a presynaptic GABA agonist, has been shown to be effective in children and adolescents with dystonia, but again, no controlled trials of oral baclofen for the treatment of CD have been conducted.38
Mexiletine, an oral derivative of lidocaine, has been found effective for the treatment of CD.39,40 In patients with CD refractory to BoNT-A and oral pharmacologic treatment, riluzole demonstrated improvement in symptoms.41 However, future better-designed studies are needed before these drugs can be recommended as a routine treatment for CD.
Botulinum Neurotoxin
Botulinum neurotoxin (BoNT), which inhibits the release of acetylcholine from the presynaptic nerve terminal, thereby causing local muscular weakness, is the most commonly used pharmacotherapy for the treatment of dystonia. Seven serotypes of BoNT (A, B, C, D, E, F, and G) are known. OnabotulinumtoxinA (Botox®), abobotulinumtoxinA (Dysport®), incobotulinumtoxinA (Xeomin®), and rimabotulinumtoxinB (Myobloc®/Neurobloc®) are different BoNT products currently used in the treatment of CD.42,43
In patients with CD, injections of BoNT reduce or eliminate involuntary muscle activity and improve control of neck movement, pain, and range of motion. An evidence-based review concluded that BoNT should be offered as an option for the treatment of CD (Level A), which is the highest level of recommendation.44 BoNT therapy is considered the most effective approach to CD, demonstrating improvement in up to 75% of patients within 1 week of injection and with duration of effect lasting an average of 12 weeks.45
Evidence-based systematic reviews have concluded that BoNT therapy is safe and effective for the treatment of CD.44,46 A large number of controlled and open-label trials of BoNT for the treatment of CD have demonstrated both subjective and objective improvements in symptoms and in pain relief.47-50 Even though there is no evidence that BoNT slows the progression of the disease, this treatment has favorably altered the natural history. Early intervention has eliminated many of the long-term complications of CD, such as contractures and radiculopathy, and has demonstrated meaningful QOL improvements.47
The long-term efficacy and safety of BoNT has been evaluated in several studies. Several longitudinal studies have shown that a substantial benefit from repeat BoNT injections can be maintained for several years and even decades.51-53
Adverse events associated with the use of BoNT are generally mild, dose dependent, infrequent, and self-limited. The most commonly reported adverse events include dysphagia, neck weakness, and local pain at the injection site.47,54 Dizziness, dry mouth, a flu-like syndrome, lethargy, dysphonia, and generalized weakness have also been reported. In a meta-analysis, approximately 25% of patients treated with onabotulinumtoxinA reported mild-to-moderate adverse events compared with 15% of patients in the control group (P < .001).46 Focal weakness was the only adverse event that occurred significantly more often in patients treated with onabotulinumtoxinA compared with controls. The results of a systematic review and analysis of published literature indicate differences in adverse-event rates between BoNT preparations. Even though the various type A BoNT preparations seem to have a similar efficacy and adverse effect profile, rimabotulinumtoxinB tends to be associated with a higher frequency of dry mouth in the majority of studies.49,55-57 Dysphagia, the most feared side effect of BoNT treatment of CD, is more likely to occur when bilateral sternocleidomastoid or scalene muscles are injected in patients with anterocollis, but even this type of CD can be safely and effectively treated when an appropriate dose of BoNT and injection technique is applied.43
The results of various analyses indicate that there are differences in clinical efficacy, adverse events, and immunoresistance between the different BpNT preparations and that the doses are not interchangeable.42,58 A guiding therapeutic principle of BoNT injection is to achieve optimal results with the lowest possible dose and frequency of administration. This strategy is critical in order to minimize the risk of immunoresistance.59 Although the frequency of antibodies directed against BoNT type A toxins is relatively low (probably less than 2%), several studies have suggested that the risk of developing immunoresistance is higher with rimabotulinumtoxinB.54,60 Because of the risk of immunoresistance, it is generally recommended that the inter-injection interval not be any shorter than 3 months. It is possible, however, that preparations with lower protein loading may have lower antigenicity and therefore, may allow for more frequent injections.42
Optimal clinical outcomes with the fewest adverse events in the treatment of CD with BoNT therapy are achieved by targeting the most involved dystonic muscles, injecting a sufficient quantity of toxin, and minimizing diffusion to uninvolved muscles. The administration of BoNT requires specialized skills and a detailed understanding of the pharmacology of the product, and knowledge of the structural and functional anatomy of the affected area. Dosing and injection sites vary with clinical presentation, and physicians should provide a dose sufficient enough to weaken the dystonic muscles but minimize diffusion into adjacent, uninvolved muscles. Movement patterns may be complex and vary over time; therefore, the injection site and dosage may need to be adjusted. Electromyography (EMG) guidance may be used, particularly by less experienced clinicians, to ensure that the target muscle is injected.61
The approach to the treatment of CD with BoNT involves the following62:
• Establishment of treatment goals and realistic patient expectations
• Assessment and documentation of the severity of CD
• Identification of possible cervical contractures
• Identification and treatment of comorbid conditions that may contribute to pain
• Determination of appropriate dosing
• Selection of appropriate muscle for injection sites (52 muscles are involved in cervical movement)
• Selection of appropriate muscles based on the patient's pain and location of painful area(s)
• Further consideration of dose and concentration for each muscle
• Possible administration into multiple sites within each muscle
• Follow-up regarding benefits and/or adverse events
Dosing and Injection Technique in Patients with CD
As there is currently no agreement on an optimal concentration, dilution of BoNT varies considerably. Some suggested starting doses for BoNT treatment of patients with CD are provided in Table 1.63-65
Table 1. Suggested Starting Doses for BoNTs in Treatment of Cervical Dystonia63-65
| Muscle | Starting Dose (units) | ||
|
|
OnabotulinumtoxinAa (Average Dose)63 (Botox U) |
AbobotulinumtoxinAb (Average Dose)64 (Dysport U) |
RimabotulinumtoxinBc (Dose Range)65 (Myobloc U) |
| Trapezius | 50 | 110 | 1000–2500 |
| Sternocleidomastoid (SCM) | 40 | 130 | 1000–2500 |
| Splenius capitis | 50 | 200 | 1000–2500 |
| Levator scapulae | 50 | 110 | 1000–2500 |
| Scalene complex | 30 | 120 | 500–1000 |
U= Units
aThe dose should be reduced by 50% if both SCM muscles are injected.
bAverage dose in double-blind pivotal phase 3 studies combined. The recommended starting dose of abobotulinumtoxinA for the treatment of CD
is 500 units given IM as a divided dose among affected muscles in patients with or without a history of prior treatment with BoNT.
cRecommended initial dose for patients with a prior history of tolerating BoNT is 2500 to 5000 units divided among affected muscles. A lower
starting dose is recommended for patients without a prior history of tolerating BoNT injections. Consider a dose reduction of 50% for each muscle
when injecting SCM or scalene muscles.
Surgical Management
Surgical intervention in dystonia currently includes a variety of procedures: intrathecal baclofen pump implantation, selective peripheral denervation, and GPi DBS. As none of these procedures provide consistent or reliable relief, they are considered only in patients with CD who continue to be disabled despite optimal medical therapy, including BoNT chemodenervation. Because of limited success with the other procedures, GPi DBS is gradually becoming the surgical treatment of choice in patients with cranial-cervical dystonia.66-69 Several authors have noted that patients with phasic CD respond better to GPi DBS than those with the tonic form of dystonia, and lower-than-usual stimulation frequencies (60–100 Hz) may be associated with more robust improvements.70 DBS is considered relatively safe in the hands of experienced neurosurgeons; the most serious complications include hemorrhagic stroke (1% per brain hemisphere), infection (5% requiring surgery for hardware removal), and lead fracture (1% per year).66 In a prospective, multicenter, 3-year follow-up of patients with generalized dystonia, bilateral GPi DBS was found to provide sustained motor benefit.71 Long-term positive clinical outcomes have been reported for up to 8 years in patients with generalized dystonia,72 but well-designed controlled and longitudinal studies are needed to gain a better understanding of the role of DBS in the treatment of CD.>
Physical Therapy
No randomized controlled trials have been conducted to examine the effects of physical therapy intervention for the treatment of CD. With the exception of a small study of EMG biofeedback,73 no large case series or controlled trials examining the benefit of any other techniques have been published. A limited number of case reports and class B studies suggest that physical therapy in conjunction with BoNT treatment may extend treatment efficacy in CD.74,75
Summary
CD is the most common form of focal dystonia and has a significant impact on the QOL of those with the condition. Although the pathogenesis of CD remains largely speculative, significant progress has been made toward defining the clinical characteristics of CD. Current management of CD is predominantly based on medical and surgical approaches. Further investigation of BoNT for clinical use and novel treatment strategies may represent a significant advance in the treatment of focal dystonia.
V. SPASMODIC DYSPHONIA
Spasmodic dysphonia (SD), also known as laryngeal dystonia, is a disorder of central motor processing that causes action-induced laryngeal muscular spasms. SD is a focal form of dystonia, causing interruptions of speech and affecting voice quality. Symptoms develop gradually over months to years, can be exacerbated by stress, and disappear during nonlinguistic tasks, such as laughing, singing, and sighing.
a) Epidemiology
According to the National Spasmodic Dysphonia Association (NSDA), an estimated 50,000 persons in North America are affected by SD, but the actual number may be higher due to ongoing misdiagnosis or undiagnosed cases of the disorder.76 Women are more commonly affected by SD, and the average age of onset is 45 years.77 Rarely, childhood viral illness has been associated with SD. A significantly higher incidence of both essential tremor and writer's cramp was found in patients with SD, thus providing evidence that SD is a form of focal dystonia. In a small proportion of patients, the condition may progress to another part of the body.<
The 2 major subtypes of SD are adductor SD, which is caused by irregular hyperadduction of the vocal folds, and abductor SD, caused by spasms of the posterior cricoarytenoid (PCA) muscle that result in abduction of the vocal folds.78
Adductor SD is much more common. Patients with adductor SD exhibit a choked, strained-strangled voice quality, with abrupt initiation and termination, resulting in short breaks in phonation. The voice is generally reduced in loudness and sounds monotonal. Vocal tremor is frequently observed, along with a slow speech rate and decreased smoothness of speech. Speech intelligibility is generally decreased. Patients with abductor SD exhibit a breathy, effortful voice quality with abrupt termination, resulting in low-volume whispered speech.
The degree of voice impairment in SD varies widely according to the muscles involved, the severity of muscle hyperactivity, and the patient's compensatory mechanisms. Symptoms may be exacerbated by stress.79 The psychosocial effects of the condition include straining or severing ties with family and friends, increased risk of depression and anxiety, and impairment of job performance and limitation on career advancement.80
Diagnosing SD requires a comprehensive examination that includes a patient's medical and family history, as well as a detailed neurological and head and neck examination, with particular attention to any spasms, dysfunction, or tremor. Fiberoptic laryngoscopy can be performed with or without a video recording to observe glottal function and detect disruptions, spasms, breathy breaks, and tremor during speech. A consultation by a speech pathologist should be undertaken to make acoustic and aerodynamic measurements and to evaluate for tremor, frequency, pitch, amplitude perturbation, harshness, fluency breaks, and breathiness during speech. A stroboscopic examination or percutaneous laryngeal EMG can help define the frequency and regularity of tremor, if present. The patient's report of difficult words or sounds is important.81
d) Management of SD
Although there is no cure for SD, medical and surgical approaches to the treatment of SD have been aimed at chemodenervation of the involved laryngeal muscles with BoNT chemodenervation. Surgical procedures, although still occasionally used, are often associated with permanent hoarseness and other complications.82-84
An evidence-based review concluded that onabotulinumtoxinA should be considered as a treatment option for adductor SD (Level B).44 Unilateral injections into the thyroarytenoid muscles appear to be more optimal compared with bilateral injections,85 but no consensus has been reached on which approach is best. There is insufficient evidence to support or refute the use of BoNT in abductor SD. OnabotulinumtoxinA treatment in the abductor patient is more difficult and is associated with greater risks, including mild to severe stridor caused by PCA paralysis.81 Among older patients, abductor SD is also associated with a poorer treatment outcome.79,81 In a study of 901 patients with SD (747 patients with adductor SD and 154 patients with abductor SD), treatment with onabotulinumtoxinA resulted in an average 90% improvement in function for patients with adductor SD and 66.7% improvement for patients with abductor SD.78
In a long-term single-blind study of patients with SD who were treated with serial onabotulinumtoxinA injections for up to 2 years, voice recordings revealed significant improvement over baseline at all time points and sustained responsiveness to therapy over time, based on overall symptom severity.86 In addition, outcome assessments of mental health and social functioning clearly showed onabotulinumtoxinA brought about statistically significant improvements for patients with SD. Treatment with onabotulinumtoxinA also markedly lessened the patient's perception of dysphonia.87
For some patients with adductor SD who found little or no improvement after onabotulinumtoxinA injection into the thyroarytenoid muscles, targeting the lateral cricoarytenoid muscles has resulted in an improved voice.87 Some patients with adductor SD have a persistence of strained/strangled voice even after BoNT injection of the thyroarytenoid relieves the hyperadduction. In such patients, additional injection of the supraglottic portion of the lateral cricoarytenoid muscles through an EMG-guided thyrohyoid approach corrects the compensatory or supraglottic hyperadduction.88 The use of fine-wire EMG to precisely determine the dystonic activity of the affected muscles has led to targeted therapy individualized for each patient and also revealed the involvement of the interarytenoid muscle in some cases of SD.87
OnabotulinumtoxinA therapy for SD appears to be well tolerated, without severe adverse effects. Mild postinjection paralytic dysphonia with hoarseness is the most commonly reported adverse effect in patients with adductor SD.79,81 In a retrospective 6-year case series review of patients with abductor SD who were treated with simultaneous bilateral onabotulinumtoxinA, injections into the PCA muscle have been shown to be safe.89 There was, however, a 5% incidence of significant dyspnea and a 2% incidence of dysphagia.89 In an open-label, dose-finding study evaluating the treatment of adductor SD, rimabotulinumtoxinB was found to be safe and effective based on the patient's rating of the change in the severity of spasms.89 Breathiness was the most common side effect, but was mild in intensity and of short duration in patients treated with rimabotulinumtoxinB injections.
The dosage of BoNT used in SD varies depending on the toxin preparation and the injection technique used (see Table 2).48,91 In a 1-year study that compared onabotulinumtoxinA with rimabotulinumtoxinB in the treatment of adductor SD, both preparations were shown to be safe and effective. RimabotulinumtoxinB was associated with a more rapid onset of action (2.09 days vs 3.2 days; P = .028) and a shorter duration of action (10.8 weeks vs 17 weeks; P = .002). The safety profiles for both toxin preparations were comparable.92
Table 2. Suggested Doses (Units) for Botulinum Toxins in Treatment of Spasmodic Dysphonia91
| Diagnosis and Treatment Technique | OnabotulinumtoxinA (Botox U) |
AbobotulinumtoxinA (Dysport U) |
RimabotulinumtoxinB (Myobloc U) |
| ADSD unilateral injections | 5–15 | 15–45 | 250–500 |
| ADSD bilateral injections | 0.5–3 | 1.5–9 | 100–250 |
| ABSD unilateral injections | 15 | 45 | NDA |
| ABSD bilateral injections | 1.25–1.75 | 4.5–6 | NDA |
| Vocal tremor | 2.5 | 7.5 | 100–250 |
| Laryngeal spasmodic dyspnea | 2.5 | 7.5 | 100–250 |
Abbreviations:
U= Units
ABSD= abductor spasmodic dysphonia
ADSD= adductor spasmodic dysphonia
NDA= no dose currently available
No randomized controlled trials have examined the effects of speech therapy for the treatment of SD. Based on class B studies, speech therapy in conjunction with BoNT injections may have beneficial effects in patients with SD.44,74,75
Summary
SD—a focal form of dystonia caused by laryngeal muscle spasms that result in interruptions of speech—affects voice quality and significantly impacts QOL. The pathogenesis of SD remains largely speculative. Current management of SD consists predominantly of BoNT injections. Further investigation of BoNT for clinical use and novel treatment strategies may represent a significant advance in the treatment of this focal dystonia.
VI. FOCAL HAND DYSTONIA — Writer's Cramp
Focal hand dystonias are relatively common, although accurate epidemiological data are lacking. Focal hand dystonia can be related to an occupational activity, such as writing, certain sports activities, or playing a musical instrument.1 Hair stylists, musicians, court reporters, athletes, and others who work repetitively with their hands are most commonly affected. Although the etiology of most task-specific dystonias is not known, peripheral injury occasionally precedes the onset of some task-specific dystonias93 Brain imaging studies are usually unremarkable, but subtle white matter abnormalities have been reported in patients with writer's cramp (WC),94 suggesting that reduced cerebellothalamic connectivity results in loss of inhibition and increased motor activation responses at the cortical level. Since the most common task-specific dystonia is WC, the discussion will focus on on this form of focal dystonia.95
a) Epidemiology
The prevalence of WC is estimated to be 14 per million persons, according to the ESDE Collaborative Group.2 WC is found predominantly in patients of middle age and has been defined as a task-specific dystonia triggered by writing, with no impairment to any other motor task.96-98 A history of trauma has not been associated with WC.97 Primary focal dystonias, such as CD and blepharospasm, affect women more than men; however, WC is twice as common among men.99
A current hypothesis suggests reduced intracortical inhibition is involved in WC. Two-dimensional magnetic resonance spectroscopy has revealed decreased GABA levels in the sensorimotor cortex and lentiform nuclei contralateral to the affected hand in patients with WC.100 Although families with WC are relatively rare, genetics is increasingly recognized to play a role in patients with task-specific dystonias, including WC and musician's dystonia.101
WC is characterized by an abnormally tight grip while writing, often accompanied by flexion of the wrist and abduction of the shoulder with elevation of the elbow. Other abnormal dystonic patterns in WC include extension of the wrist, flexion-pronation and supination of the forearm, flexion and extension of the fingers, extension and forced flexion of the thumb and index finger, or shoulder elevation.96
Focal Hand Dystonia, specifically writer’s cramp, demonstrating abnormal excessive, and progressive, flexion of the fingers while writing
Video courtesy of Mark Hallett, MD with permission from patient
The motor abnormality usually starts insidiously and tends to progress before stabilizing. Symptoms may be painless or accompanied by painful hand and forearm cramping.102 Tremor, which may involve 1 or both hands, can occur at rest or during the performance of the task. Tension and discomfort in the fingers and the forearm are common; in most patients, pain is not present.97 Spontaneous remissions of WC occur in only about 5% of cases.
The physiologic abnormalities and muscles affected vary among patients with hand cramps.103 Overflow contraction is seen in muscles not usually activated in a given task, along with failure of other needed muscles to be activated.104 Although WC rarely evolves into segmental or generalized dystonia, overflow to the opposite side can occur. Furthermore, voluntary movement of the unaffected hand often triggers dystonia in the affected, resting hand—so-called "mirror" dystonia—even without performance of a particular task.105 The use of EMG in affected individuals has identified muscle co-contraction, with a loss of the normal alternating pattern of agonist and antagonist muscles, prolonged muscle bursts, and loss of reciprocal inhibition.103,106
Diagnosis of WC includes a comprehensive history of the patient's complaint and routine physical and neurological examination. The affected hand(s) may appear normal when examined and may perform normally on other tasks, even those that activate the same muscles.95 The task of identifying the specific muscles involved may be made difficult by the presence of voluntary compensatory movements. The patient should therefore be carefully examined at rest and during movements that activate the dystonia. Examination for mirror movements in the affected and unaffected hands is useful in clinically characterizing WC and in planning future therapy, particularly in selecting the most appropriate muscles for BoNT injection.105 Selection of muscles for injection should be based on the clinical evaluation, patient report of local discomfort or tightness, and/or EMG evidence of excessive muscle activation.
d) Management of WC
Currently available interventions for WC include sensorimotor training, immobilization, rehabilitative therapies, oral medications, BoNT injections, and GPi DBS. No single treatment strategy is appropriate for all cases of WC. One of the most effective methods is adapting tasks in an attempt to avoid triggering the dystonic movements—a strategy that may involve occupational therapy or using assistive/adaptive devices.107 However, the long-term benefits of these nonpharmacologic approaches have not been established. Oral medications, such as dopamine agonists and antagonists, baclofen, benzodiazepines, and anticholinergics produce little or no benefit, and adverse effects usually limit the usefulness of these drugs.
BoNT injections have become a mainstay of therapy in the management of WC. An evidence-based review concluded that BoNT should be offered as a treatment option for WC (Level B).44 More than minimal improvement was demonstrated in 80% of patients with WC after receiving at least 1 injection series of onabotulinumtoxinA.108 Benefits began approximately 1 week after injection, reaching peak improvement at 2 weeks and lasting approximately 3 months.95,108 In a prospective study of 47 patients, subjective benefit was reported in 73% of patients with WC treated with onabotulinumtoxinA. Patients with a pronation/flexion pattern of dystonia showed the best and the most sustained improvement with BoNT. However, the presence of dystonic tremor was predictive of poor response to BoNT treatment.96
The primary objective of the treatment of WC is to optimize function by relaxing the abnormally contracting muscles without inducing excessive weakness while improving motor function106 Weakness of the target muscle is the most common side effect of BoNT treatment, but most cases have been mild and transient.108,109
Despite the clinical improvement provided by onabotulinumtoxinA for treatment of WC, a substantial proportion of patients do not continue injections for more than 2 years. This rate of discontinuation has been attributed to an unacceptable trade-off between disability from the dystonia and the weakness brought on by repeated BoNT injections, including loss of speed and coordination.104 Many patients are not entirely satisfied with the degree of benefit provided by BoNT and in some, the treatment must be supplemented with anticholinergic drugs, muscle relaxants, and physical therapy.
The use of BoNT to treat WC requires thoughtful technique, including customization of dose and muscle selection (see Figure 1 and Table 3).108 Many clinicians advocate EMG or nerve stimulation guidance to optimize needle localization for injection; however, further data are needed to establish this recommendation.
Figure 1. Muscles of Forearm Commonly Involved in Focal Hand DystoniaTable 3. Doses of Botulinum Toxins for
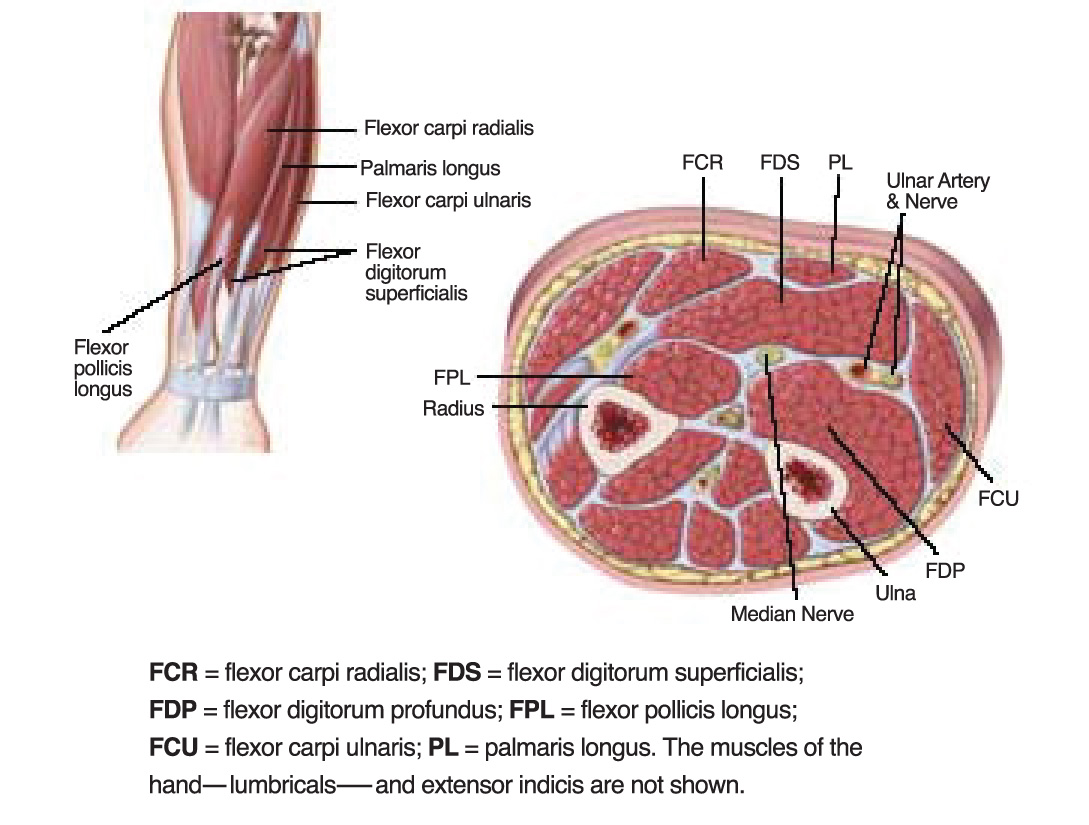
| Muscle | OnabotulinumtoxinA
(Botox U) |
AbobotulinumtoxinA
(Dysport U) |
| Flexor digitorum profundus | 20–40 | 60–120 |
| Flexor carpi ulnaris | ||
| Flexor digitorum superficialis | 25–50 | 75–150 |
| Flexor carpi radialis | ||
| Flexor pollicis longus | 10–20 | 30–50 |
| Extensor pollicis longus | ||
| Pronator teres | 20–30 | 60–100 |
| Lumbricals/extensor indicis | 5–10 | 15–30 |
| Extensor digitorum communis | 15–25 | 50–75 |
U= Units
Thalamic and GPi DBS have replaced ablative procedures (eg, thalamotomy) as the surgical treatments of patients with task-specific dystonias refractory to BoNT and medical therapies. 110-112
Summary
WC is the most common form of task-specific dystonia. The pathogenesis of WC remains largely speculative, but genetic predisposition may play a role. Current management consists primarily of BoNT injections. Further investigation of BoNT for clinical use and novel treatment strategies may represent a significant advance in the treatment of this focal dystonia.
RETURN TO TOPREFERENCES
- 1. Jankovic J, Ashoori A. Movement disorders in musicians. Mov Disord. 2008;14:1957–1965.
- 2. Warner TT. A prevalence study of primary dystonia in eight European countries. Epidemiological Study of Dystonia in Europe (ESDE) Collaborative Group. J Neurol. 2000;247:787–792.
- 3. Jankovic J, Tsui J, Bergeron C. Prevalence of cervical dystonia and spasmodic torticollis in the United States general population. Parkinsonism Relat Disord. 2007;13:411–416.
- 4. Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–234.
- 5. Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol. 2009;8:441–446.
- 6. Schwarz CS, Bressman SB. Genetics and treatment of dystonia. Neurol Clin. 2009;27:697-718, vi.
- 7. de Carvalho Aguiar PM, Ozelius LJ. Classification and genetics of dystonia. Lancet Neurol. 2002;1:316–325.
- 8. Albanese A, Barnes MP, Bhatia KP, et al. A systematic review on the diagnosis and treatment of primary (idiopathic) dystonia and dystonia plus syndromes: report of an EFNS/MDS-ES Task Force. Eur J Neurol. 2006;13:433–444.
- 9. Singer C, Velickovic M. Cervical dystonia: etiology and pathophysiology. Neurol Clin. 2008;26 (suppl 1):9–22.
- 10. Bressman S. Genetics of dystonia. J Neural Transm Suppl. 2006;(70):489–495.
- 11. Kock N, Naismith TV, Boston HE, et al. Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum Mol Genet. 2006;15:1355–1364.
- 12. Opal P, Tintner R, Jankovic J, et al. Intrafamilial phenotypic variability of the DYT1 dystonia: from asymptomatic TOR1A gene carrier status to dystonic storm. Mov Disord. 2002;17:339–345.
- 13. Bressman SB, Sabatti C, Raymond D, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology. 2000;54:1746–1752.
- 14. Bressman SB, Heiman GA, Nygaard TG, et al. A study of idiopathic torsion dystonia in a non-Jewish family: evidence for genetic heterogeneity. Neurology. 1994;44:283–287.
- 15. Bressman SB. Dystonia genotypes, phenotypes, and classification. Adv Neurol. 2004;94:101–107.
- 16. Yang JF, Wu T, Li JY, Li YJ, Zhang YL, Chan P. DYT1 mutations in early onset primary torsion dystonia and Parkinson disease patients in Chinese populations. Neurosci Lett. 2009;450:117–121.
- 17. Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23.
- 18. Schneider SA, Bhatia KP, Hardy J. Complicated recessive dystonia parkinsonism syndromes. Mov Disord. 2009;24:490–499.
- 19. Albanese A, Lalli S. Is this dystonia? Mov Disord. 2009;24:1725–1731.
- 20. National Spasmodic Torticollis Association. Spasmodic Torticollis/Cervical Dystonia. 2010. http://www.torticollis.org/. Accessed April 28, 2010.
- 21. Defazio G, Abbruzzese G, Livrea P, Berardelli A. Epidemiology of primary dystonia. Lancet Neurol. 2004;3:673–678.
- 22. Sex-related influences on the frequency and age of onset of primary dystonia. Epidemiologic Study of Dystonia in Europe (ESDE) Collaborative Group. Neurology. 1999;53:1871–1873.
- 23. Bloom F, Squire LR, Spitzer N, Gage F, Albright T, eds. Encyclopedia of Neuroscience. Philadelphia, PA: Elsevier Science; 2009.
- 24. Camfield L, Ben-Shlomo Y, Warner TT. Impact of cervical dystonia on quality of life. Mov Disord. 2002;17:838–841.
- 25. Pekmezovic T, Svetel M, Ivanovic N, et al. Quality of life in patients with focal dystonia. Clin Neurol Neurosurg. 2009;111:161–164.
- 26. Fabbrini G, Berardelli I, Moretti G, et al. Psychiatric disorders in adult-onset focal dystonia: a case-control study. Mov Disord. 2010;25:459–465.
- 27. Gundel H, Wolf A, Xidara V, Busch R, Ceballos-Baumann AO. Social phobia in spasmodic torticollis. J Neurol Neurosurg Psychiatry. 2001;71:499–504.
- 28. Jahanshahi M. Factors that ameliorate or aggravate spasmodic torticollis. J Neurol Neurosurg Psychiatry. 2000;68:227–229.
- 29. Consky ES, Lang AE. Clinical assessments of patients with cervical dystonia. In: Jankovic J, Hallett M, eds. Therapy with Botulinum Toxin. New York, NY: Marcel Dekker, Inc; 1994:211–237.
- 30. Tsui JK, Eisen A, Stoessl AJ, Calne S, Calne DB. Double-blind study of botulinum toxin in spasmodic torticollis. Lancet. 1986;2(8501):245–247.
- 31. Odergren T, Tollback A, Borg J. Efficacy of botulinum toxin for cervical dystonia: a comparison of methods for evaluation. Scand J Rehabil Med. 1994;26:191–195.
- 32. Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scale for the primary torsion dystonias. Neurology. 1985;35:73–77.
- 33. Cano SJ, Warner TT, Linacre JM, et al. Capturing the true burden of dystonia on patients: the Cervical Dystonia Impact Profile (CDIP-58). Neurology. 2004;63:1629–1633.
- 34. Muller J, Wissel J, Kemmler G, et al. Craniocervical dystonia questionnaire (CDQ-24): development and validation of a disease-specific quality of life instrument. J Neurol Neurosurg Psychiatry. 2004;75:749–753.
- 35. Jankovic J. Treatment of dystonia. Lancet Neurol. 2006;5:864–872.
- 36. Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol. 2009;8:844–856.
- 37. Adler CH, Kumar R. Pharmacological and surgical options for the treatment of cervical dystonia. Neurology. 2000;55(12)(suppl 5):S9–S14.
- 38. Greene PE, Fahn S. Baclofen in the treatment of idiopathic dystonia in children. Mov Disord. 1992;7:48–52.
- 39. Lucetti C, Nuti A, Gambaccini G, et al. Mexiletine in the treatment of torticollis and generalized dystonia. Clin Neuropharmacol. 2000;23:186–189.
- 40. Ohara S, Hayashi R, Momoi H, Miki J, Yanagisawa N. Mexiletine in the treatment of spasmodic torticollis. Mov Disord. 1998;13:934–940.
- 41. Muller J, Wenning GK, Wissel J, et al. Riluzole therapy in cervical dystonia. Mov Disord. 2002;17:198–200.
- 42. Albanese A, Bigalke H, Foster K, et al. Basic and therapeutic aspects of botulinum and tetanus toxins. Toxicon. 2009;54:549–708.
- 43. Jankovic J. Disease-oriented approach to botulinum toxin use. Toxicon. 2009;54:614–623.
- 44. Simpson DM, Blitzer A, Brashear A, et al. Assessment: Botulinum neurotoxin for the treatment of movement disorders (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;70:1699–1706.
- 45. Dauer WT, Burke RE, Greene P, Fahn S. Current concepts on the clinical features, aetiology and management of idiopathic cervical dystonia. Brain. 1998;121(pt 4):547–560.
- 46. Naumann M, Jankovic J. Safety of botulinum toxin type A: a systematic review and meta-analysis. Curr Med Res Opin. 2004;20:981–990.
- 47. Jankovic J. Treatment of cervical dystonia with botulinum toxin. Mov Disord. 2004;19(suppl 8):S109–S115.
- 48. Truong D, Duane DD, Jankovic J, et al. Efficacy and safety of botulinum type A toxin (Dysport) in cervical dystonia: results of the first US randomized, double-blind, placebo-controlled study. Mov Disord. 2005;20:783–791.
- 49. Pappert EJ, Germanson T, for The Myobloc/Neurobloc European Cervical Dystonia Study Group. Botulinum toxin Type B vs. Type A in toxin-naive patients with cervical dystonia: randomized, double-blind, noninferiority trial. Mov Disord. 2008;23:510–517.
- 50. Truong D, Brodsky M, Lew M, et al, on behalf of the Global Dysport Cervical Dystonia Study Group. Long-term efficacy and safety of botulinum toxin type A (Dysport) in cervical dystonia. Parkinsonism Relat Disord. 2010;16:316–323.
- 51. Hsiung GY, Das SK, Ranawaya R, Lafontaine AL, Suchowersky O. Long-term efficacy of botulinum toxin A in treatment of various movement disorders over a 10-year period. Mov Disord. 2002;17:1288–1293.
- 52. Haussermann P, Marczoch S, Klinger C, Landgrebe M, Conrad B, Ceballos-Baumann A. Long-term follow-up of cervical dystonia patients treated with botulinum toxin A. Mov Disord. 2004;19:303–308.
- 53. Mejia NI, Vuong KD, Jankovic J. Long-term botulinum toxin efficacy, safety and immunogenicity. Mov Disord. 2005;20:592–597.
- 54. Jankovic J, Hunter C, Dolimbek BZ, et al. Clinico-immunologic aspects of botulinum toxin type B treatment of cervical dystonia. Neurology. 2006;67:2233–2235.
- 55. Comella CL, Jankovic J, Shannon KM, et al; Dystonia Study Group. Comparison of botulinum toxin serotypes A and B for the treatment of cervical dystonia. Neurology. 2005;65:1423–1429.
- 56. Tintner R, Gross R, Winzer UF, Smalky KA, Jankovic J. Autonomic function after botulinum toxin type A or B: a double-blind, randomized trial. Neurology. 2005;65:765–767.
- 57. Dressler D, Comella CL. Comparative clinical trials of botulinum toxins. In: Jankovic J, Albanese A, Atassi MZ, Dolly JO, Hallett M, Mayer N, eds. Botulinum and Other Neurotoxins: Translating Science Into Therapeutic Applications. Philadelphia, PA: Butterworth-Heinemann (Elsevier); , 2009:398–405.
- 58. Chapman MA, Barron R, Tanis DC, Gill CE, Charles PD. Comparison of botulinum neurotoxin preparations for the treatment of cervical dystonia. Clin Ther. 2007;29:1325–1337.
- 59. Tintner R, Jankovic J. Botulinum toxin for the treatment of cervical dystonia. Expert Opin Pharmacother. 2001;2:1985–1994.
- 60. Berman B, Seeberger L, Kumar R. Long-term safety, efficacy, dosing, and development of resistance with botulinum toxin type B in cervical dystonia. Mov Disord. 2005;20:233–237.
- 61. Comella CL, Jankovic J, Brin MF. Use of botulinum toxin type A in the treatment of cervical dystonia. Neurology. 2000;55(12) (suppl 5):S15–S21.
- 62. Hallett M, Jankovic J, Silberstein SD, eds. A Multispecialty Guide to the Use of Botulinum Neurotoxins. Rev. ed. Irvine CA: University of California, Irvine; New York, NY: The Neurotoxin Institute; 2006.
- 63. Brashear A, Mayer N. Dosing and administration of botulinum toxin for muscle overactivity in adults with an upper motor neuron syndrome. In: Spasticity and Other Forms of Muscle Overactivity in the Upper Motor Neuron Syndrome. WE MOVE; 2008:207–218.
- 64. Dysport (abobotulinumtoxinA) prescribing information. Wrexham, UK: Ipsen Biopharm Ltd.
- 65. Myobloc Dosing and Injection—Insights From Clinical Trials: Dosing and Injection Sites. http://solstice.mdtrainer.com/Flash.aspx. Accessed May 19, 2010.
- 66. Ostrem JL, Starr PA. Treatment of dystonia with deep brain stimulation. Neurotherapeutics. 2008;5:320–330.
- 67. Jeong SG, Lee MK, Kang JY, Jun SM, Lee WH, Ghang CG. Pallidal deep brain stimulation in primary cervical dystonia with phasic type: clinical outcome and postoperative course. J Korean Neurosurg Soc. 2009;46:346–350.
- 68. Moro E, Piboolnurak P, Arenovich T, Hung SW, Poon YY, Lozano AM. Pallidal stimulation in cervical dystonia: clinical implications of acute changes in stimulation parameters. Eur J Neurol. 2009;16:506–512.
- 69. Pretto TE, Dalvi A, Kang UJ, Penn RD. A prospective blinded evaluation of deep brain stimulation for the treatment of secondary dystonia and primary torticollis syndromes. J Neurosurg. 2008;109:405–409.
- 70. Alterman RL, Shils JL, Miravite J, Tagliati M. Lower stimulation frequency can enhance tolerability and efficacy of pallidal deep brain stimulation for dystonia. Mov Disord. 2007;22:366–368.
- 71. Vidailhet M, Vercueil L, Houeto JL, et al. Bilateral, pallidal, deep-brain stimulation in primary generalised dystonia: a prospective 3 year follow-up study. Lancet Neurol. 2007;6:223-9.
- 72. Isaias IU, Alterman RL, Tagliati M. Deep brain stimulation for primary generalized dystonia: long-term outcomes. Arch Neurol. 2009;66:465–470.
- 73. Smania N, Corato E, Tinazzi M, Montagnana B, Fiaschi A, Aglioti SM. The effect of two different rehabilitation treatments in cervical dystonia: preliminary results in four patients. Funct Neurol. 2003;18:219–225.
- 74. Delnooz CC, Horstink MW, Tijssen MA, van de Warrenburg BP. Paramedical treatment in primary dystonia: a systematic review. Mov Disord. 2009;24:2187–2198.
- 75. Swope D, Barbano R. Treatment recommendations and practical applications of botulinum toxin treatment of cervical dystonia. Neurol Clin. 2008;26(suppl 1):54–65.
- 76. National Spasmodic Dysphonia Association. Frequently asked questions. http://www.dysphonia.org/faq.asp?nav=pub#q.5. Accessed May 19, 2010.
- 77. Schweinfurth JM, Billante M, Courey MS. Risk factors and demographics in patients with spasmodic dysphonia. Laryngoscope. 2002;112:220–223.
- 78. Blitzer A, Brin MF, Stewart CF. Botulinum toxin management of spasmodic dysphonia (laryngeal dystonia): a 12-year experience in more than 900 patients. Laryngoscope. 1998;108:1435–1441.
- 79. Tisch SH, Brake HM, Law M, Cole IE, Darveniza P. Spasmodic dysphonia: clinical features and effects of botulinum toxin therapy in 169 patients—an Australian experience. J Clin Neurosci. 2003;10:434–438.
- 80. Smith E, Taylor M, Mendoza M, Barkmeier J, Lemke J, Hoffman H. Spasmodic dysphonia and vocal fold paralysis: outcomes of voice problems on work-related functioning. J Voice. 1998;12:223–232.
- 81. Gibbs SR, Blitzer A. Botulinum toxin for the treatment of spasmodic dysphonia. Otolaryngol Clin North Am. 2000;33:879–894.
- 82. Chhetri DK, Berke GS. Treatment of adductor spasmodic dysphonia with selective laryngeal adductor denervation and reinnervation surgery. Otolaryngol Clin North Am. 2006;39:101–109.
- 83. Ludlow CL. Treatment for spasmodic dysphonia: limitations of current approaches. Curr Opin Otolaryngol Head Neck Surg. 2009;17:160–165.
- 84. Nakamura K, Muta H, Watanabe Y, Mochizuki R, Yoshida T, Suzuki M. Surgical treatment for adductor spasmodic dysphonia—efficacy of bilateral thyroarytenoid myectomy under microlaryngoscopy. Acta Otolaryngol. 2008;128:1348–1353.
- 85. Bielamowicz S, Stager SV, Badillo A, Godlewski A. Unilateral versus bilateral injections of botulinum toxin in patients with adductor spasmodic dysphonia. J Voice. 2002;16:117–123.
- 86. Damrose JF, Goldman SN, Groessl EJ, Orloff LA. The impact of long-term botulinum toxin injections on symptom severity in patients with spasmodic dysphonia. J Voice. 2004;18:415–422.
- 87.Hillel AD, Maronian NC, Waugh PF, Robinson L, Klotz DA. Treatment of the interarytenoid muscle with botulinum toxin for laryngeal dystonia. Ann Otol Rhinol Laryngol. 2004;113:341–348.
- 88. Young N, Blitzer A. Management of supraglottic squeeze in adductor spasmodic dysphonia: a new technique. Laryngoscope. 2007;117:2082–2084.
- 89. Stong BC, DelGaudio JM, Hapner ER, Johns MM III. Safety of simultaneous bilateral botulinum toxin injections for abductor spasmodic dysphonia. Arch Otolaryngol Head Neck Surg. 2005;131:793–795.
- 90. Adler CH, Bansberg SF, Krein-Jones K, Hentz JG. Safety and efficacy of botulinum toxin type B (Myobloc) in adductor spasmodic dysphonia. Mov Disord. 2004;19:1075–1079.
- 91. Truong DD, Bhidayasiri R. Botulinum toxin therapy of laryngeal muscle hyperactivity syndromes: comparing different botulinum toxin preparations. Eur J Neurol. 2006;13(suppl):36–41.
- 92. Blitzer A. Botulinum toxin A and B: a comparative dosing study for spasmodic dysphonia. Otolaryngol Head Neck Surg. 2005;133:836–838.
- 93. Jankovic J. Peripherally induced movement disorders. Neurol Clin. 2009;27:821–832.
- 94. Delmaire C, Vidailhet M, Wasserman D, et al. Diffusion abnormalities in the primary sensorimotor pathways in writer's cramp. Arch Neurol. 2009;66:502–508.
- 95. Karp BI. Botulinum toxin treatment of occupational and focal hand dystonia. Mov Disord. 2004;19(suppl 8):S116–S119.
- 96. Djebbari R, du Montcel ST, Sangla S, Vidal JS, Gallouedec G, Vidailhet M. Factors predicting improvement in motor disability in writer's cramp treated with botulinum toxin. J Neurol Neurosurg Psychiatry. 2004;75:1688–1691.
- 97. Jedynak PC, Tranchant C, de Beyl DZ. Prospective clinical study of writer's cramp. Mov Disord. 2001;16:494–499.
- 98. Marsden CD, Sheehy MP. Writer's cramp. Trends Neurosci. 1990;13:148–153.
- 99. Soland VL, Bhatia KP, Marsdan CD. Sex prevalence of focal dystonias. J Neurol Neurosurg Psychiatry. 1996;60:204–205.
- 100.Levy LM, Hallett M. Impaired brain GABA in focal dystonia. Ann Neurol. 2002;51:93–101.
- 101.Schmidt A, Jabusch HC, Altenmüller E, et al. Etiology of musician's dystonia: familial or environmental? Neurology. 2009;72:1248–1254.
- 102.Sheehy MP, Rothwell JC, Marsden CD. Writer's cramp. Adv Neurol. 1988;50:457–472.
- 103.Cohen LG, Hallett M. Hand cramps: clinical features and electromyographic patterns in a focal dystonia. Neurology. 1988;38:1005–1012.
- 104.Torres-Russotto D, Perlmutter JS. Task-specific dystonias: a review. Ann N Y Acad Sci. 2008;1142:179–199.
- 105.Sitburana O, Wu LJ, Sheffield JK, Davidson A, Jankovic J. Motor overflow and mirror dystonia. Parkinsonism Relat Disord. 2009;15:758–761.
- 106.Cohen LG, Hallett M, Geller BD, Hochberg F. Treatment of focal dystonias of the hand with botulinum toxin injections. J Neurol Neurosurg Psychiatry. 1989;52:355–363.
- 107.Tas N, Karatas GK, Sepici V. Hand orthosis as a writing aid in writer's cramp. Mov Disord. 2001;16:1185–1189.
- 108.Das CP, Dressler D, Hallett M. Botulinum toxin therapy of writer's cramp. Eur J Neurol. 2006;13 (suppl 1):55–59.
- 109.Karp BI, Cole RA, Cohen LG, Grill S, Lou JS, Hallett M. Long-term botulinum toxin treatment of focal hand dystonia. Neurology. 1994;44:70–76.
- 110.Cho CB, Park HK, Lee KJ, Rha HK. Thalamic deep brain stimulation for writer's cramp. J Korean Neurosurg Soc. 2009;46:52–55.
- 111.Fukaya C, Katayama Y, Kano T, et al. Thalamic deep brain stimulation for writer's cramp. J Neurosurgery. 2007;107:977–982.
- 112.Taira T, Hori T. Stereotactic ventrooralis thalamotomy for task-specific focal hand dystonia (writer's cramp). Stereotact Funct Neurosurg. 2003;80:88–91.


